

Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery
- PMID: 23448466
- PMCID: PMC6322545
- DOI: 10.2174/1381612811319280009
Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery
Abstract
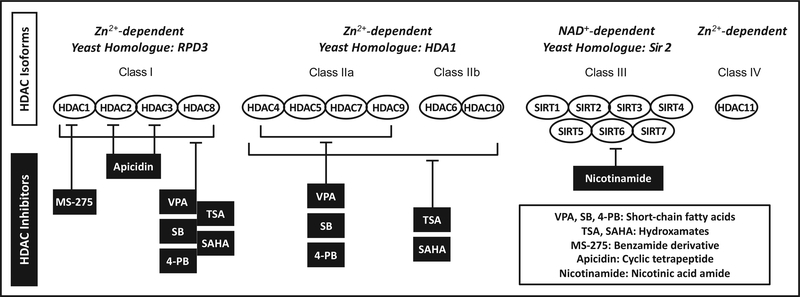
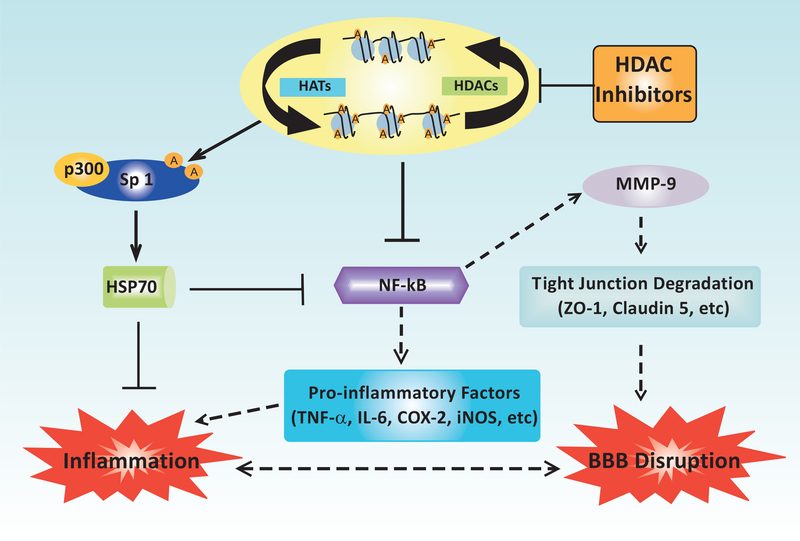
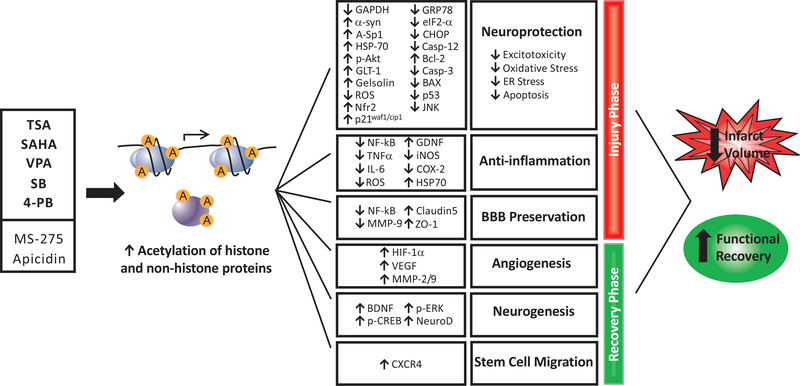
Ischemic stroke is a leading cause of death and disability worldwide, with few available treatment options. The pathophysiology of cerebral ischemia involves both early phase tissue damage, characterized by neuronal death, inflammation, and blood-brain barrier breakdown, followed by late phase neurovascular recovery. It is becoming clear that any promising treatment strategy must target multiple points in the evolution of ischemic injury to provide substantial therapeutic benefit. Histone deacetylase (HDAC) inhibitors are a class of drugs that increase the acetylation of histone and non-histone proteins to activate transcription, enhance gene expression, and modify the function of target proteins. Acetylation homeostasis is often disrupted in neurological conditions, and accumulating evidence suggests that HDAC inhibitors have robust protective properties in many preclinical models of these disorders, including ischemic stroke. Specifically, HDAC inhibitors such as trichostatin A, valproic acid, sodium butyrate, sodium 4-phenylbutyrate, and suberoylanilide hydroxamic acid have been shown to provide robust protection against excitotoxicity, oxidative stress, ER stress, apoptosis, inflammation, and bloodbrain barrier breakdown. Concurrently, these agents can also promote angiogenesis, neurogenesis and stem cell migration to dramatically reduce infarct volume and improve functional recovery after experimental cerebral ischemia. In the following review, we discuss the mechanisms by which HDAC inhibitors exert these protective effects and provide evidence for their strong potential to ultimately improve stroke outcome in patients.
Conflict of interest statement
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
Figures
Similar articles
-
Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action.J Pharmacol Exp Ther. 2007 Jun;321(3):892-901. doi: 10.1124/jpet.107.120188. Epub 2007 Mar 19. J Pharmacol Exp Ther. 2007. PMID: 17371805
-
Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage.Free Radic Biol Med. 2012 Mar 1;52(5):928-36. doi: 10.1016/j.freeradbiomed.2011.12.006. Epub 2011 Dec 17. Free Radic Biol Med. 2012. PMID: 22226832 Free PMC article.
-
Сlass II histone deacetylases in the post-stroke recovery period-expression, cellular, and subcellular localization-promising targets for neuroprotection.J Cell Biochem. 2019 Dec;120(12):19590-19609. doi: 10.1002/jcb.29266. Epub 2019 Jul 1. J Cell Biochem. 2019. PMID: 31264264
-
Updating a Strategy for Histone Deacetylases and Its Inhibitors in the Potential Treatment of Cerebral Ischemic Stroke.Dis Markers. 2020 Sep 5;2020:8820803. doi: 10.1155/2020/8820803. eCollection 2020. Dis Markers. 2020. PMID: 32963637 Free PMC article. Review.
-
[Research progress of histone deacetylase 6 inhibitors in the therapy of ischemic stroke].Sheng Li Xue Bao. 2018 Jun 25;70(3):301-309. Sheng Li Xue Bao. 2018. PMID: 29926072 Review. Chinese.
Cited by
-
Epigenetic Alterations Induced by Photothrombotic Stroke in the Rat Cerebral Cortex: Deacetylation of Histone h3, Upregulation of Histone Deacetylases and Histone Acetyltransferases.Int J Mol Sci. 2019 Jun 13;20(12):2882. doi: 10.3390/ijms20122882. Int J Mol Sci. 2019. PMID: 31200484 Free PMC article.
-
The Chemical Biology of Ferroptosis in the Central Nervous System.Cell Chem Biol. 2020 May 21;27(5):479-498. doi: 10.1016/j.chembiol.2020.03.007. Epub 2020 Apr 2. Cell Chem Biol. 2020. PMID: 32243811 Free PMC article. Review.
-
One more role for the gut: microbiota and blood brain barrier.Ann Transl Med. 2016 Jan;4(1):15. doi: 10.3978/j.issn.2305-5839.2015.10.16. Ann Transl Med. 2016. PMID: 26855951 Free PMC article. No abstract available.
-
The gut microbiota influences blood-brain barrier permeability in mice.Sci Transl Med. 2014 Nov 19;6(263):263ra158. doi: 10.1126/scitranslmed.3009759. Sci Transl Med. 2014. PMID: 25411471 Free PMC article.
-
Therapeutic effectiveness of a single exercise session combined with WalkAide functional electrical stimulation in post-stroke patients: a crossover design study.Neural Regen Res. 2021 May;16(5):805-812. doi: 10.4103/1673-5374.297078. Neural Regen Res. 2021. PMID: 33229713 Free PMC article.
References
-
- Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke 2011; 42: 2351–5. - PubMed
-
- Hajjar K, Kerr DM, Lees KR. Thrombolysis for acute ischemic stroke. J Vasc Surg 2011; 54: 901–7. - PubMed
-
- Kleindorfer D, Lindsell CJ, Brass L, Koroshetz W, Broderick JP. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke 2008; 39: 924–8. - PubMed
-
- Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 2010; 17: 197–218. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
