

Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations
- PMID: 23449933
- PMCID: PMC3687022
- DOI: 10.1158/1541-7786.MCR-12-0634-T
Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations
Abstract
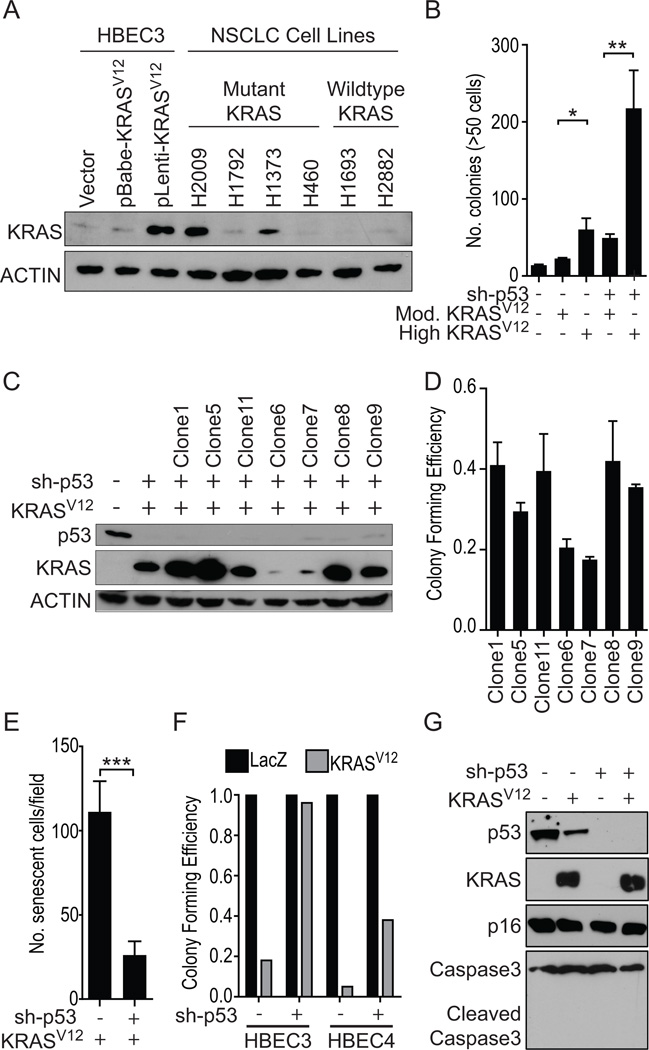
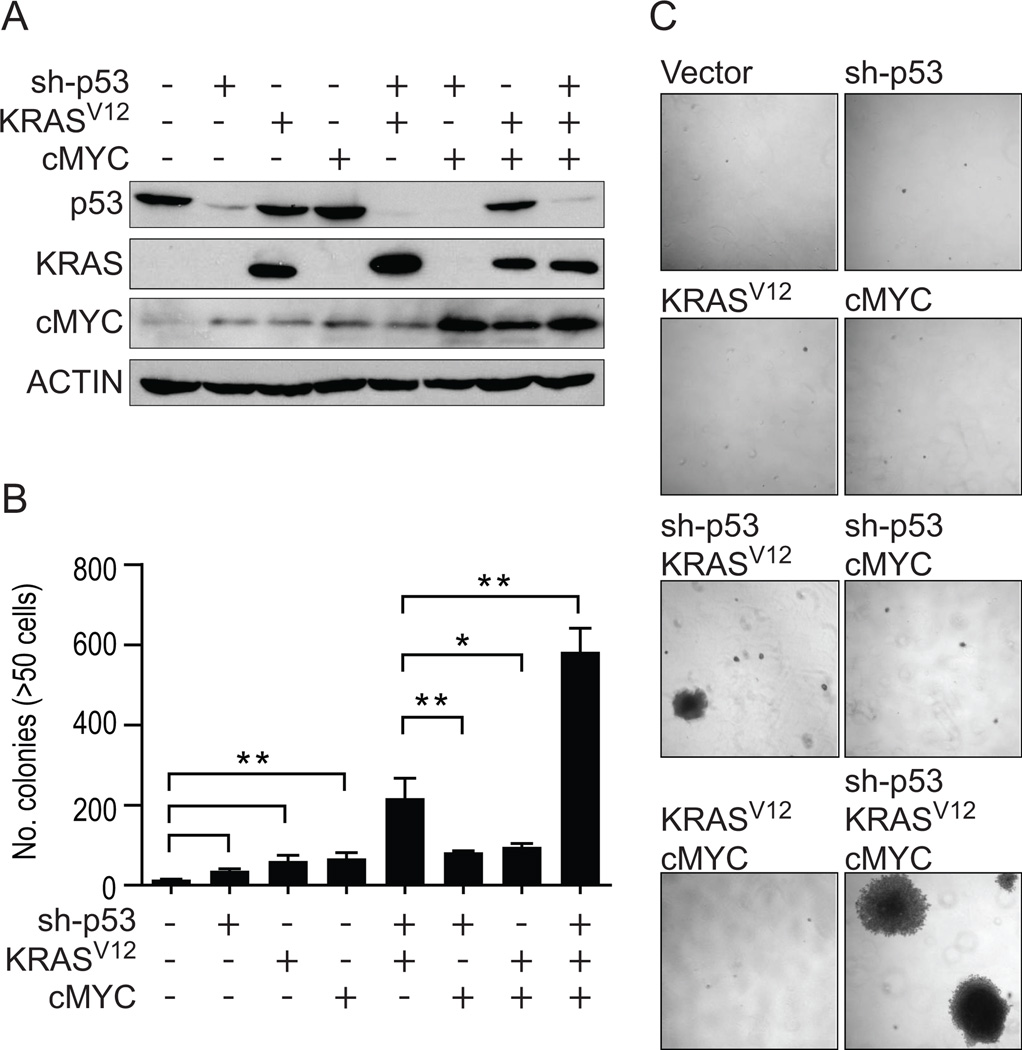
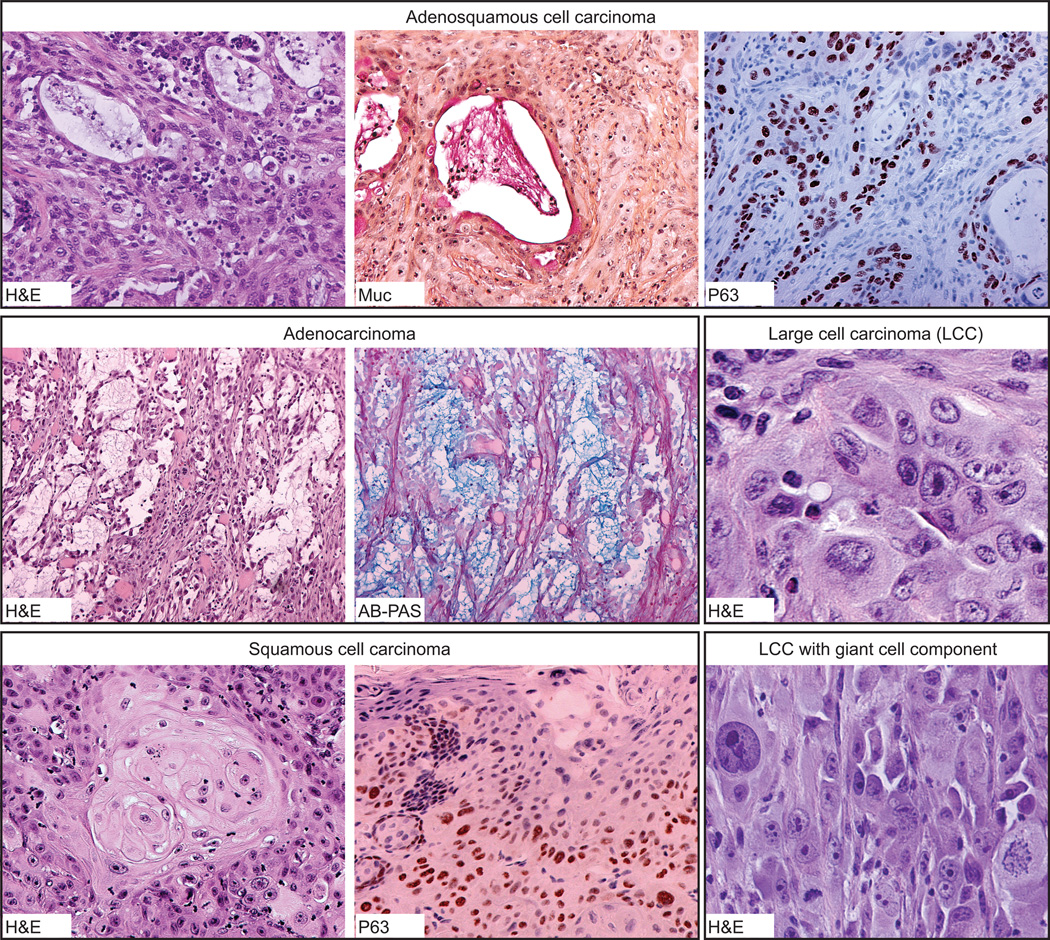
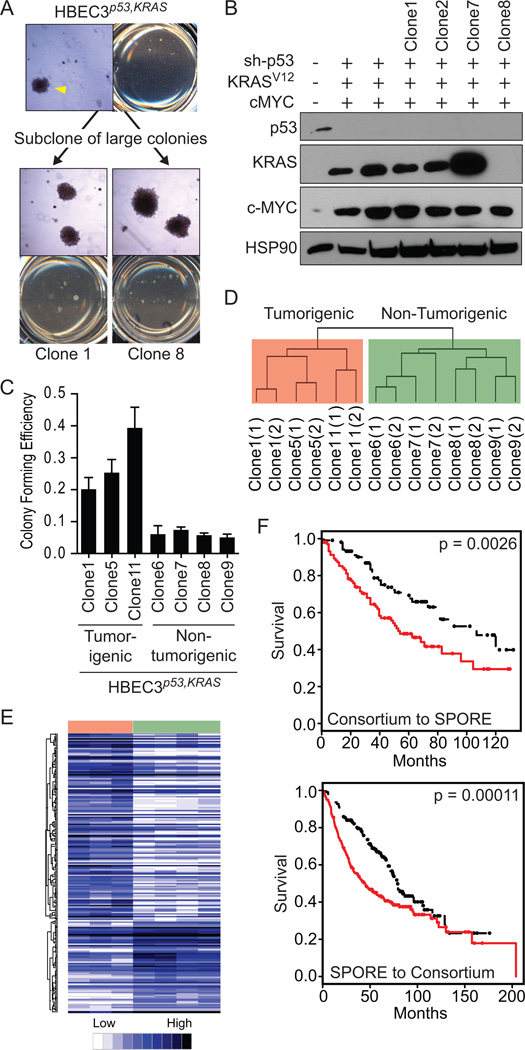
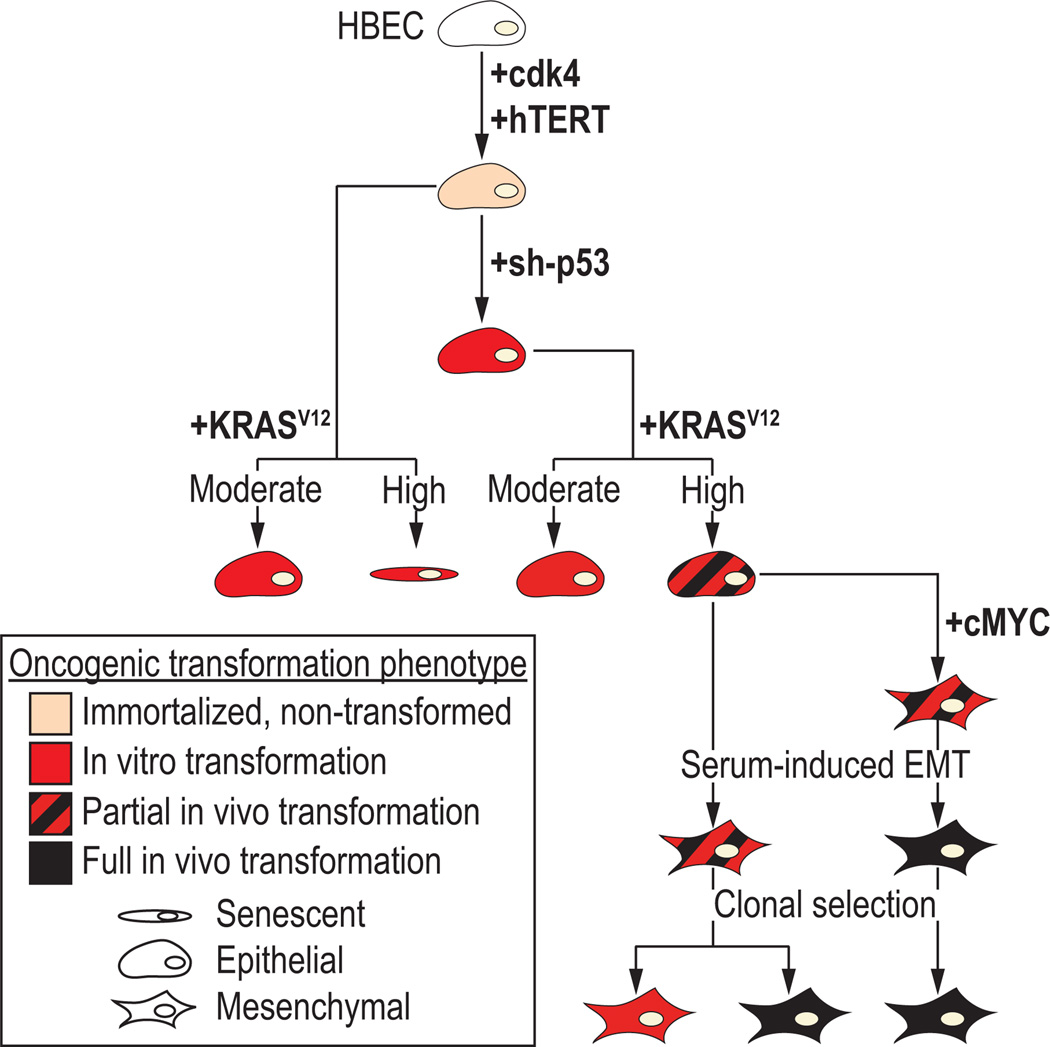
We used CDK4/hTERT-immortalized normal human bronchial epithelial cells (HBEC) from several individuals to study lung cancer pathogenesis by introducing combinations of common lung cancer oncogenic changes (p53, KRAS, and MYC) and followed the stepwise transformation of HBECs to full malignancy. This model showed that: (i) the combination of five genetic alterations (CDK4, hTERT, sh-p53, KRAS(V12), and c-MYC) is sufficient for full tumorigenic conversion of HBECs; (ii) genetically identical clones of transformed HBECs exhibit pronounced differences in tumor growth, histology, and differentiation; (iii) HBECs from different individuals vary in their sensitivity to transformation by these oncogenic manipulations; (iv) high levels of KRAS(V12) are required for full malignant transformation of HBECs, however, prior loss of p53 function is required to prevent oncogene-induced senescence; (v) overexpression of c-MYC greatly enhances malignancy but only in the context of sh-p53+KRAS(V12); (vi) growth of parental HBECs in serum-containing medium induces differentiation, whereas growth of oncogenically manipulated HBECs in serum increases in vivo tumorigenicity, decreases tumor latency, produces more undifferentiated tumors, and induces epithelial-to-mesenchymal transition (EMT); (vii) oncogenic transformation of HBECs leads to increased sensitivity to standard chemotherapy doublets; (viii) an mRNA signature derived by comparing tumorigenic versus nontumorigenic clones was predictive of outcome in patients with lung cancer. Collectively, our findings show that this HBEC model system can be used to study the effect of oncogenic mutations, their expression levels, and serum-derived environmental effects in malignant transformation, while also providing clinically translatable applications such as development of prognostic signatures and drug response phenotypes.
©2013 AACR.
Conflict of interest statement
The authors disclose no potential conflicts of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
