

Accurate identification and analysis of human mRNA isoforms using deep long read sequencing
- PMID: 23450794
- PMCID: PMC3583448
- DOI: 10.1534/g3.112.004812
Accurate identification and analysis of human mRNA isoforms using deep long read sequencing
Abstract
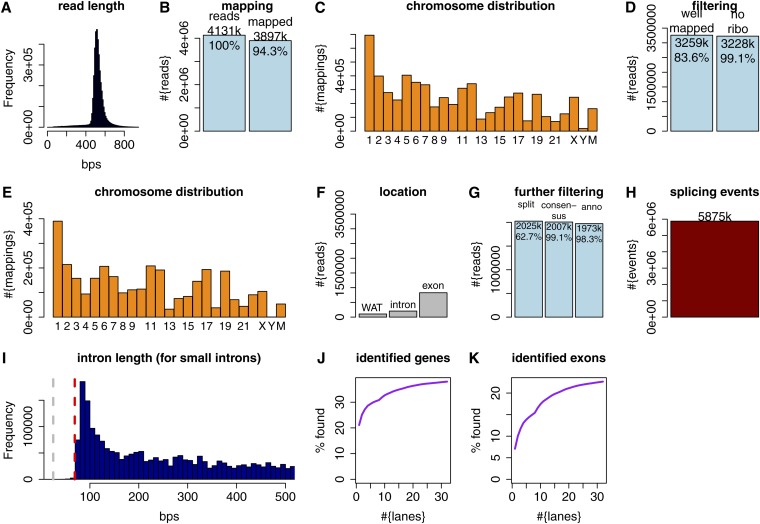
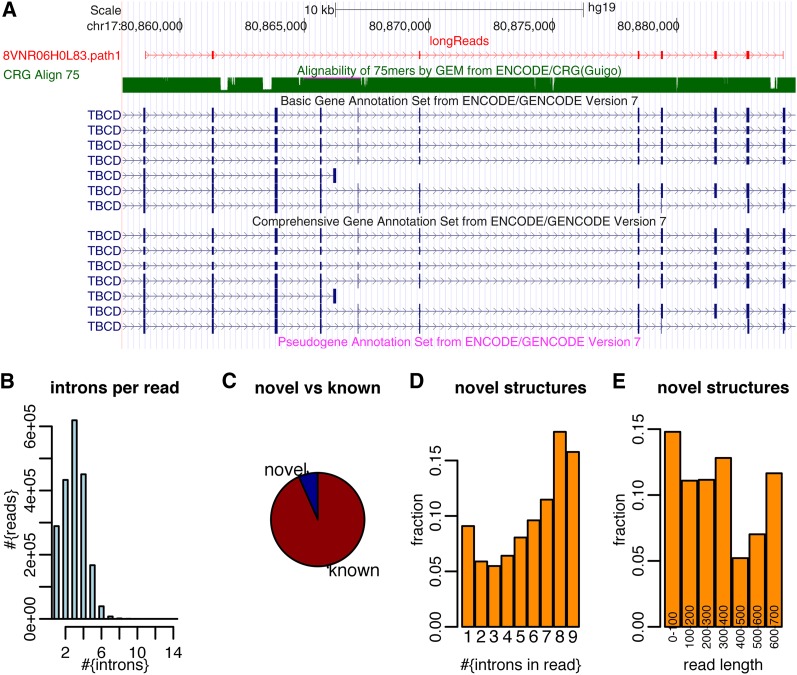
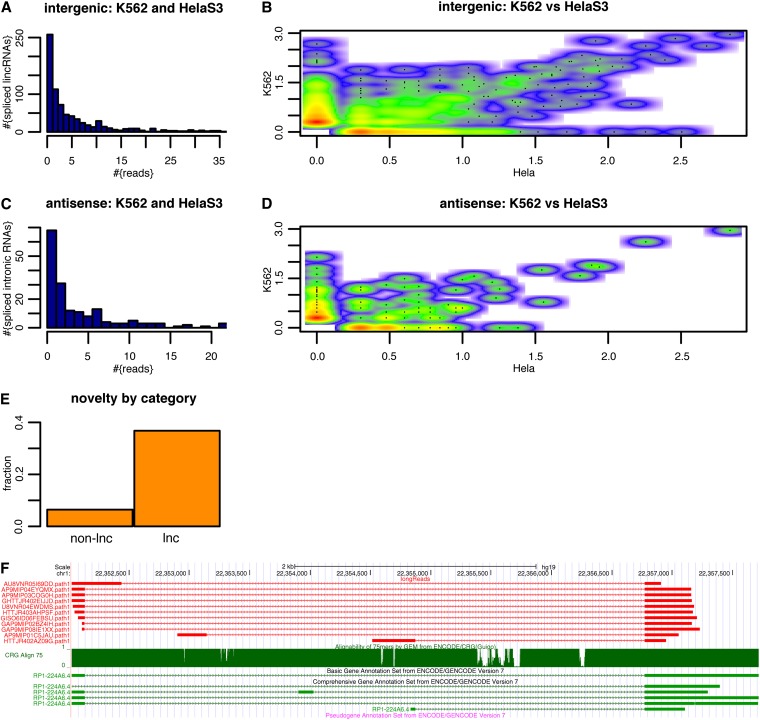
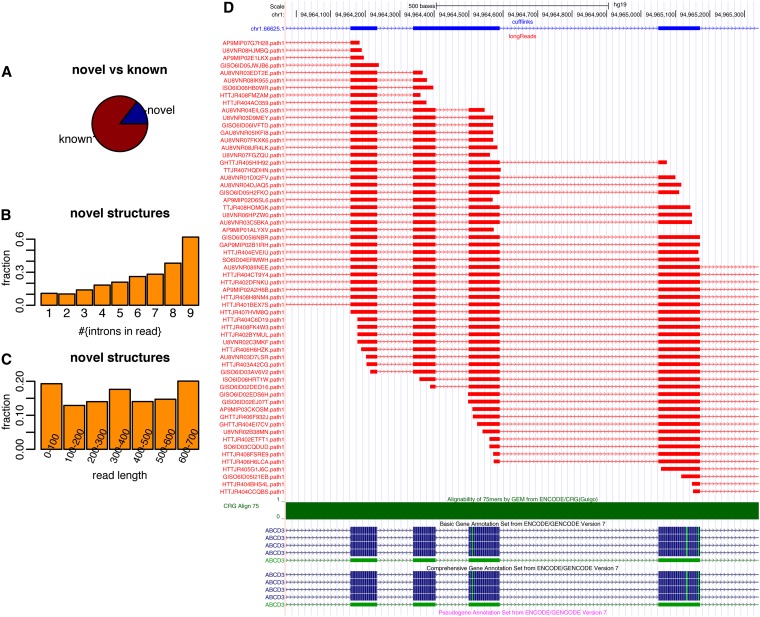
Precise identification of RNA-coding regions and transcriptomes of eukaryotes is a significant problem in biology. Currently, eukaryote transcriptomes are analyzed using deep short-read sequencing experiments of complementary DNAs. The resulting short-reads are then aligned against a genome and annotated junctions to infer biological meaning. Here we use long-read complementary DNA datasets for the analysis of a eukaryotic transcriptome and generate two large datasets in the human K562 and HeLa S3 cell lines. Both data sets comprised at least 4 million reads and had median read lengths greater than 500 bp. We show that annotation-independent alignments of these reads provide partial gene structures that are very much in-line with annotated gene structures, 15% of which have not been obtained in a previous de novo analysis of short reads. For long-noncoding RNAs (i.e., lncRNA) genes, however, we find an increased fraction of novel gene structures among our alignments. Other important aspects of transcriptome analysis, such as the description of cell type-specific splicing, can be performed in an accurate, reliable and completely annotation-free manner, making it ideal for the analysis of transcriptomes of newly sequenced genomes. Furthermore, we demonstrate that long read sequence can be assembled into full-length transcripts with considerable success. Our method is applicable to all long read sequencing technologies.
Keywords: RNA; Roche sequencing; human; splicing; transcriptome.
Figures


References
-
- Benjamini Y., Hochberg Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. B. 57: 289–300
-
- Eid J., Fehr A., Gray J., Luong K., Lyle J., et al. , 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources