

Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria
- PMID: 23453390
- PMCID: PMC3622802
- DOI: 10.1016/j.tips.2013.01.009
Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria
Abstract
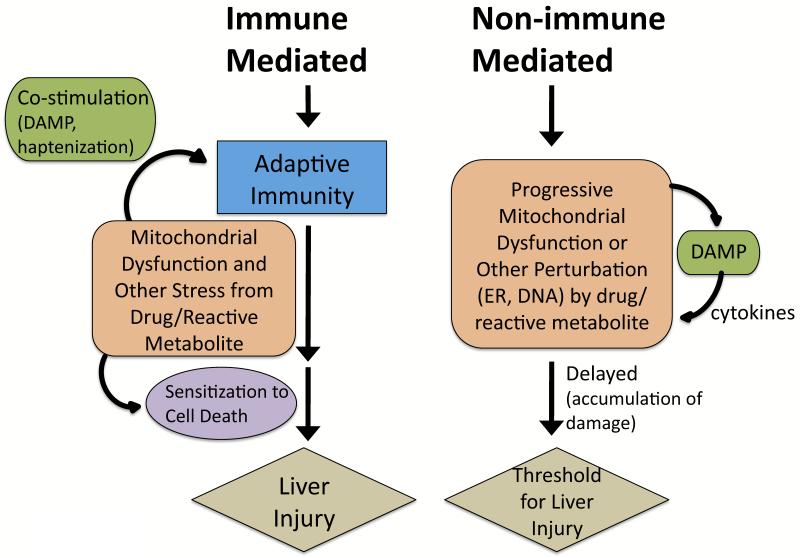
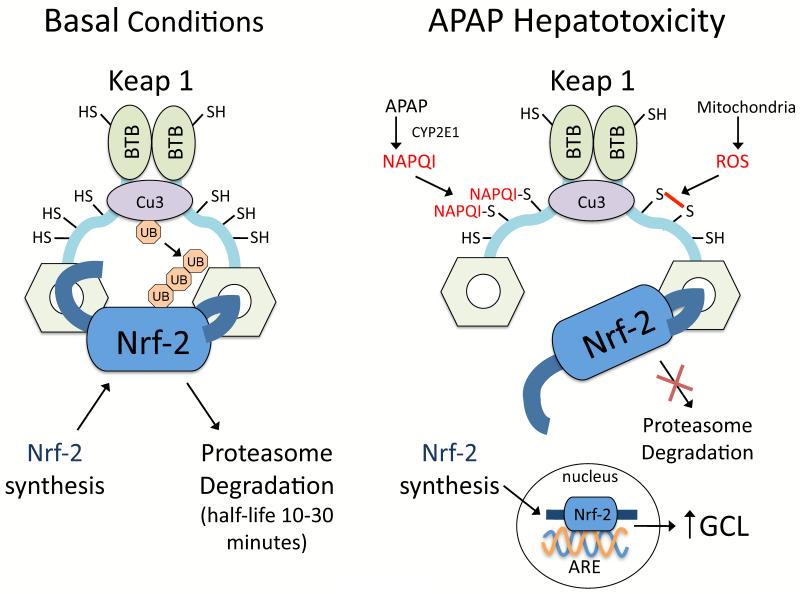
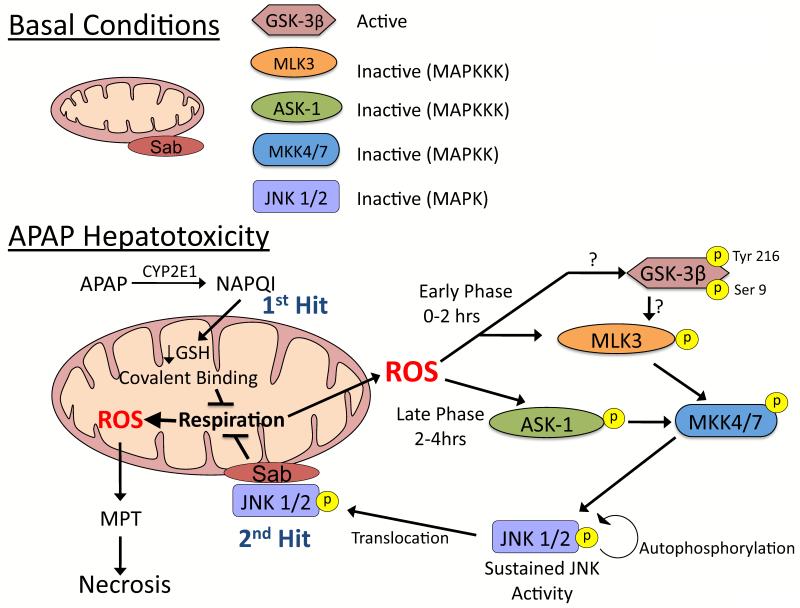
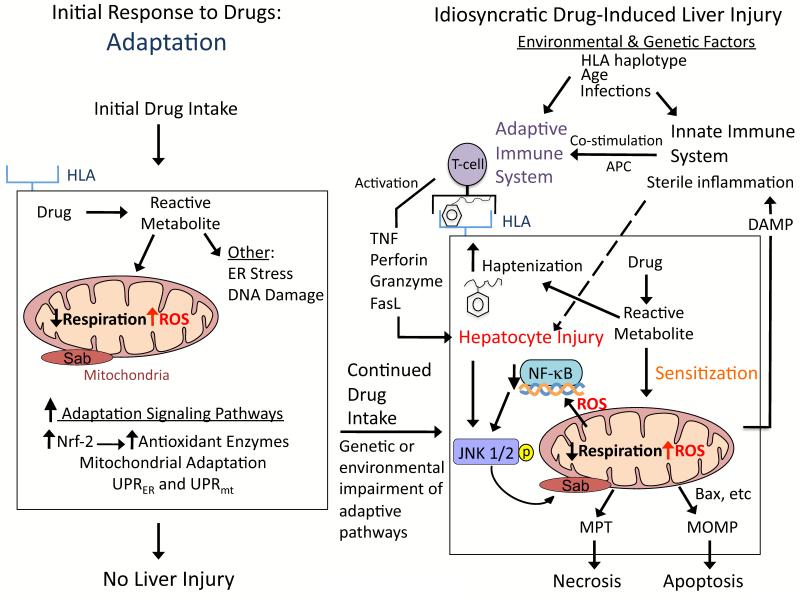
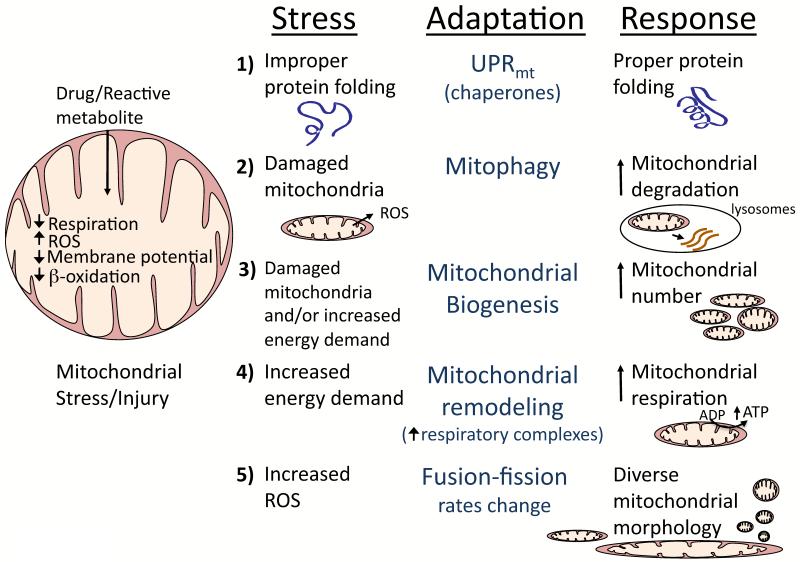
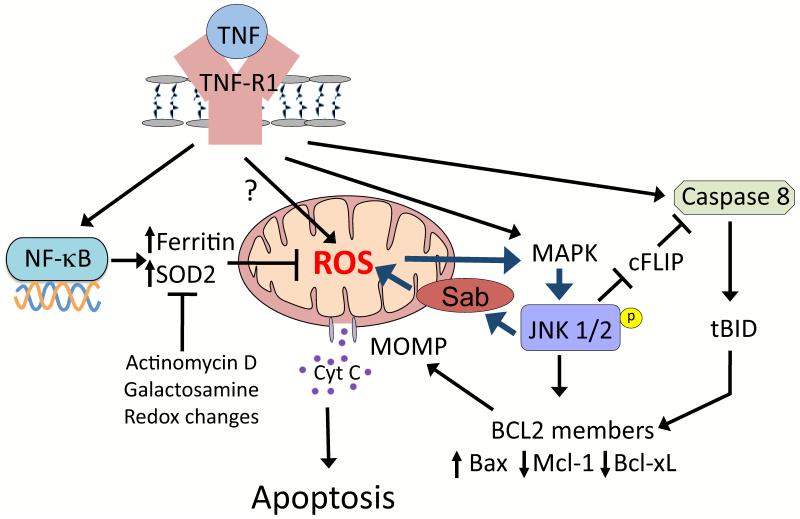
Drugs that cause liver injury often 'stress' mitochondria and activate signal transduction pathways important in determining cell survival or death. In most cases, hepatocytes adapt to the drug-induced stress by activating adaptive signaling pathways, such as mitochondrial adaptive responses and nuclear factor erythroid 2-related factor 2 (Nrf-2), a transcription factor that upregulates antioxidant defenses. Owing to adaptation, drugs alone rarely cause liver injury, with acetaminophen (APAP) being the notable exception. Drug-induced liver injury (DILI) usually involves other extrinsic factors, such as the adaptive immune system, that cause 'stressed' hepatocytes to become injured, leading to idiosyncratic DILI, the rare and unpredictable adverse drug reaction in the liver. Hepatocyte injury, due to drug and extrinsic insult, causes a second wave of signaling changes associated with adaptation, cell death, and repair. If the stress and injury reach a critical threshold, then death signaling pathways such as c-Jun N-terminal kinase (JNK) become dominant and hepatocytes enter a failsafe mode to undergo self-destruction. DILI can be seen as an active process involving recruitment of death signaling pathways that mediate cell death rather than a passive process due to overwhelming biochemical injury. In this review, we highlight the role of signal transduction pathways, which frequently involve mitochondria, in the development of DILI.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
References
-
- Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. - PubMed
-
- Holt M, Ju C. Drug-induced liver injury. Handb Exp Pharmacol. 2010:3–27. - PubMed
-
- Ulrich RG. Idiosyncratic toxicity: a convergence of risk factors. Annu Rev Med. 2007;58:17–34. - PubMed
-
- Han D, et al. Signal transduction pathways involved in drug-induced liver injury. Handb Exp Pharmacol. 2010:267–310. - PubMed
-
- Daly AK, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–819. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
