

Constitutional mutations in RTEL1 cause severe dyskeratosis congenita
- PMID: 23453664
- PMCID: PMC3591859
- DOI: 10.1016/j.ajhg.2013.02.001
Constitutional mutations in RTEL1 cause severe dyskeratosis congenita
Abstract
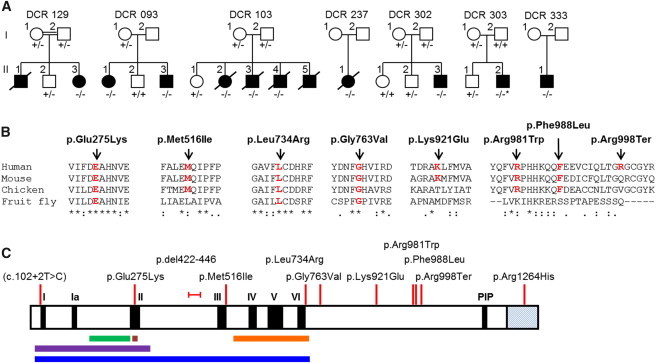
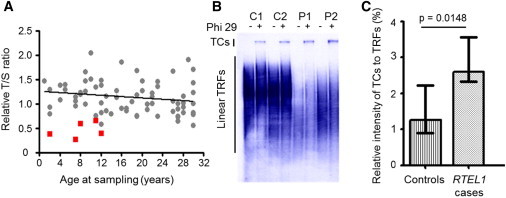
Dyskeratosis congenita (DC) and its phenotypically severe variant, Hoyeraal-Hreidarsson syndrome (HHS), are multisystem bone-marrow-failure syndromes in which the principal pathology is defective telomere maintenance. The genetic basis of many cases of DC and HHS remains unknown. Using whole-exome sequencing, we identified biallelic mutations in RTEL1, encoding a helicase essential for telomere maintenance and regulation of homologous recombination, in an individual with familial HHS. Additional screening of RTEL1 identified biallelic mutations in 6/23 index cases with HHS but none in 102 DC or DC-like cases. All 11 mutations in ten HHS individuals from seven families segregated in an autosomal-recessive manner, and telomere lengths were significantly shorter in cases than in controls (p = 0.0003). This group had significantly higher levels of telomeric circles, produced as a consequence of incorrect processing of telomere ends, than did controls (p = 0.0148). These biallelic RTEL1 mutations are responsible for a major subgroup (∼29%) of HHS. Our studies show that cells harboring these mutations have significant defects in telomere maintenance, but not in homologous recombination, and that incorrect resolution of T-loops is a mechanism for telomere shortening and disease causation in humans. They also demonstrate the severe multisystem consequences of its dysfunction.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Telomere shortening by mutations in the RTEL1 helicase cause severe form of dyskeratosis congenita, Hoyerall-Hreidarsson syndrome.Clin Genet. 2013 Sep;84(3):210. doi: 10.1111/cge.12175. Epub 2013 May 27. Clin Genet. 2013. PMID: 23621889 No abstract available.
References
-
- Dokal I. Dyskeratosis congenita. Hematology (Am Soc Hematol Educ Program) 2011;2011:480–486. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
