

Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans
- PMID: 23454694
- PMCID: PMC3712035
- DOI: 10.2337/db12-1179
Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans
Abstract
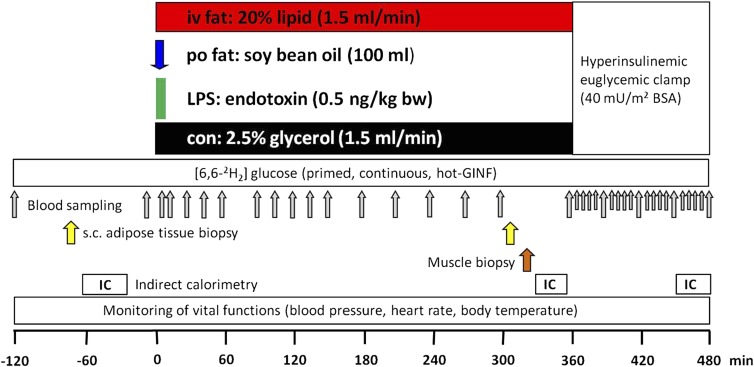
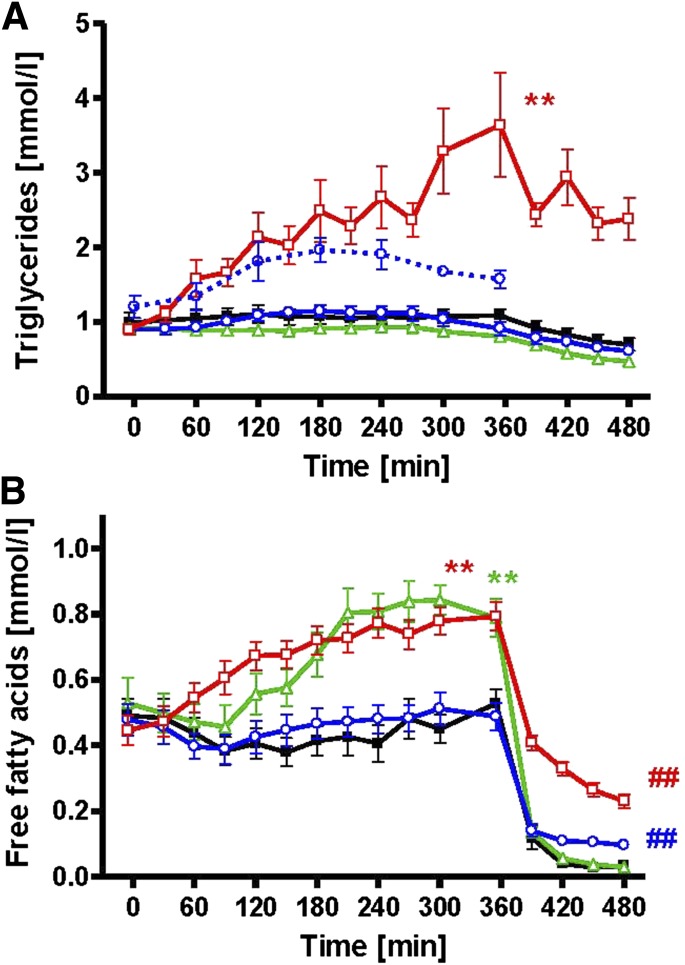
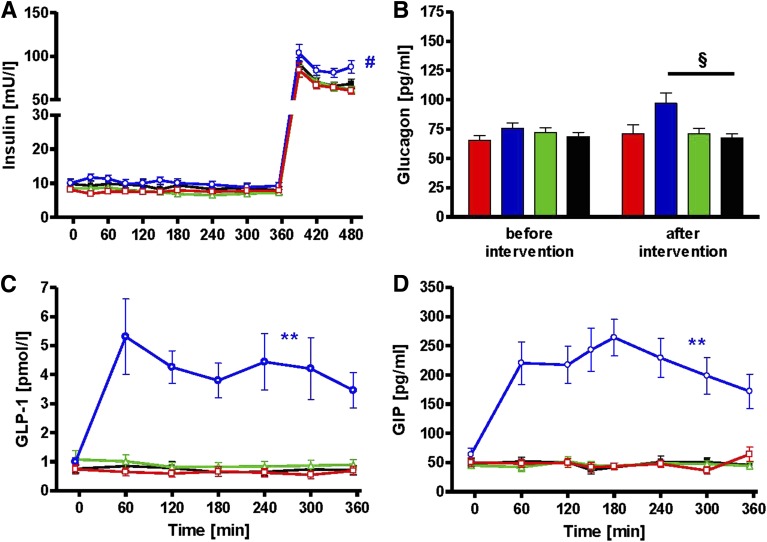
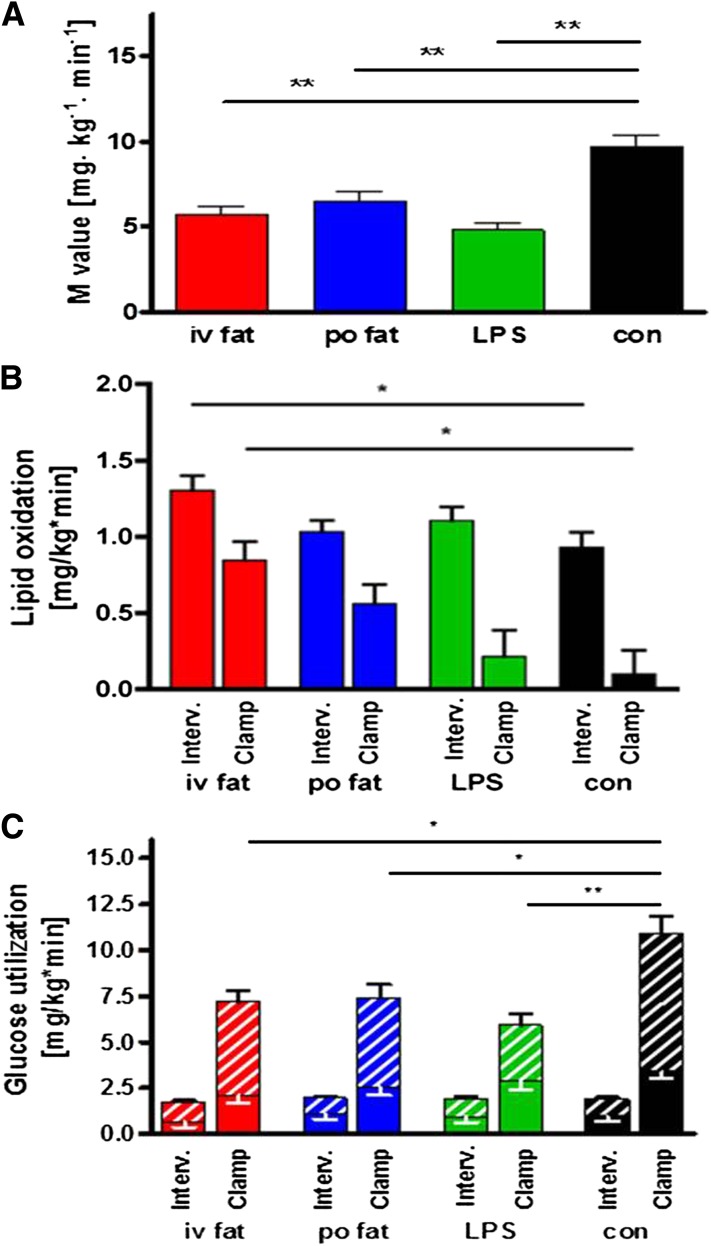
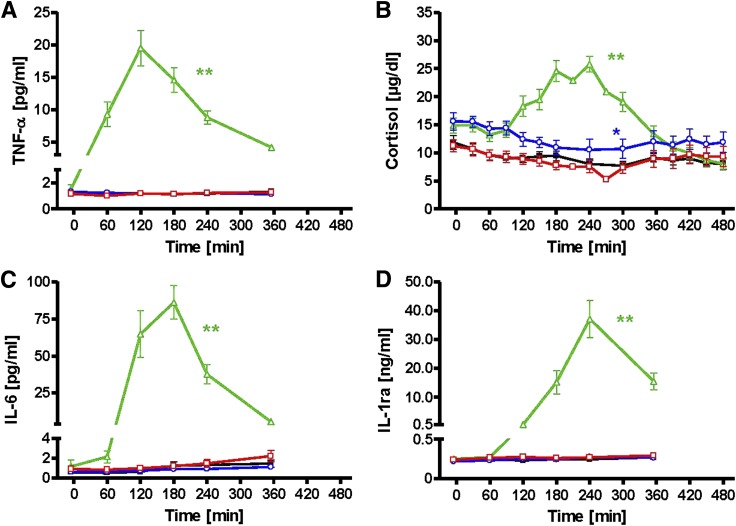
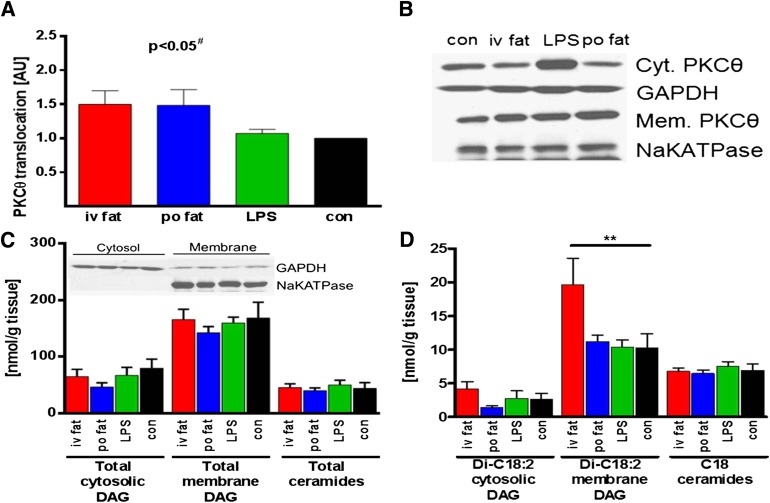
Several mechanisms, such as innate immune responses via Toll-like receptor-4, accumulation of diacylglycerols (DAG)/ceramides, and activation of protein kinase C (PKC), are considered to underlie skeletal muscle insulin resistance. In this study, we examined initial events occurring during the onset of insulin resistance upon oral high-fat loading compared with lipid and low-dose endotoxin infusion. Sixteen lean insulin-sensitive volunteers received intravenous fat (iv fat), oral fat (po fat), intravenous endotoxin (lipopolysaccharide [LPS]), and intravenous glycerol as control. After 6 h, whole-body insulin sensitivity was reduced by iv fat, po fat, and LPS to 60, 67, and 48%, respectively (all P < 0.01), which was due to decreased nonoxidative glucose utilization, while hepatic insulin sensitivity was unaffected. Muscle PKCθ activation increased by 50% after iv and po fat, membrane Di-C18:2 DAG species doubled after iv fat and correlated with PKCθ activation after po fat, whereas ceramides were unchanged. Only after LPS, circulating inflammatory markers (tumor necrosis factor-α, interleukin-6, and interleukin-1 receptor antagonist), their mRNA expression in subcutaneous adipose tissue, and circulating cortisol were elevated. Po fat ingestion rapidly induces insulin resistance by reducing nonoxidative glucose disposal, which associates with PKCθ activation and a rise in distinct myocellular membrane DAG, while endotoxin-induced insulin resistance is exclusively associated with stimulation of inflammatory pathways.
Trial registration: ClinicalTrials.gov NCT01054989.
Figures
References
-
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol 2011;29:415–445 - PubMed
-
- Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV, Ravussin E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 1995;38:1213–1217 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
