

Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS
- PMID: 23455423
- PMCID: PMC3756911
- DOI: 10.1038/nature11922
Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS
Abstract
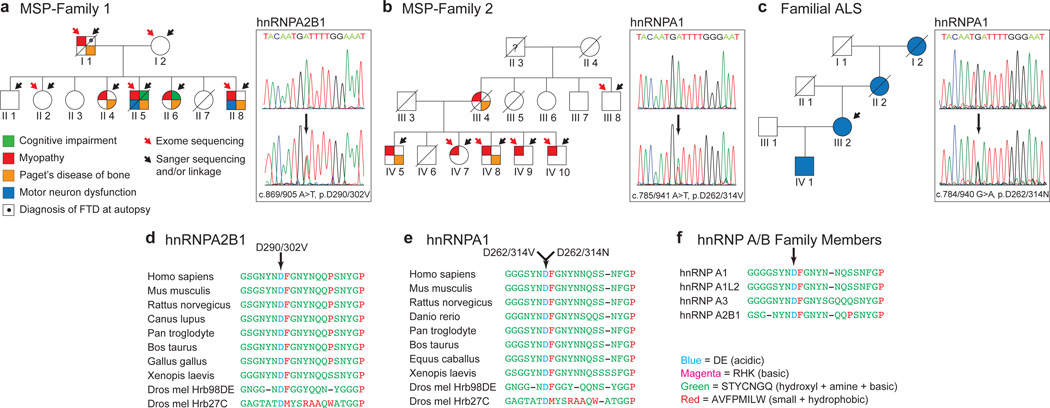
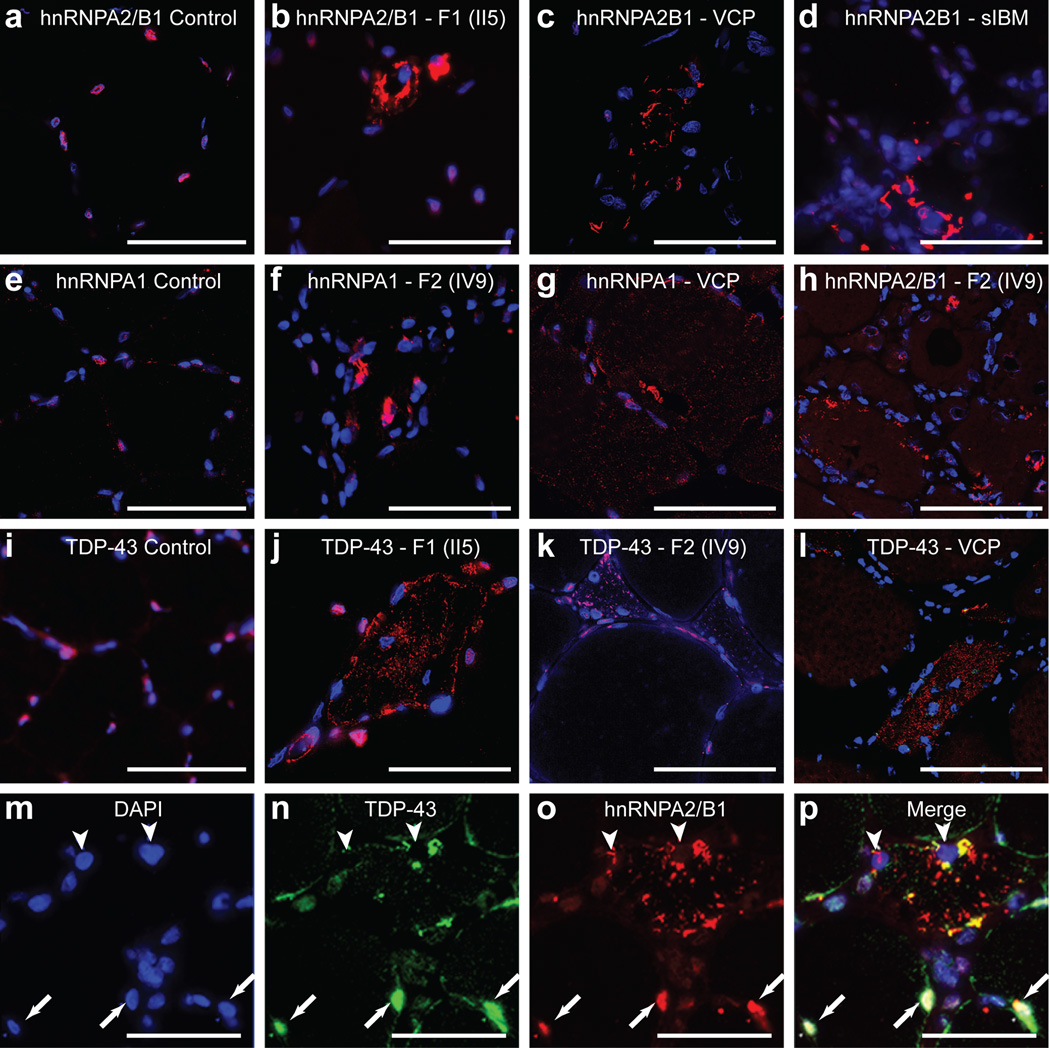
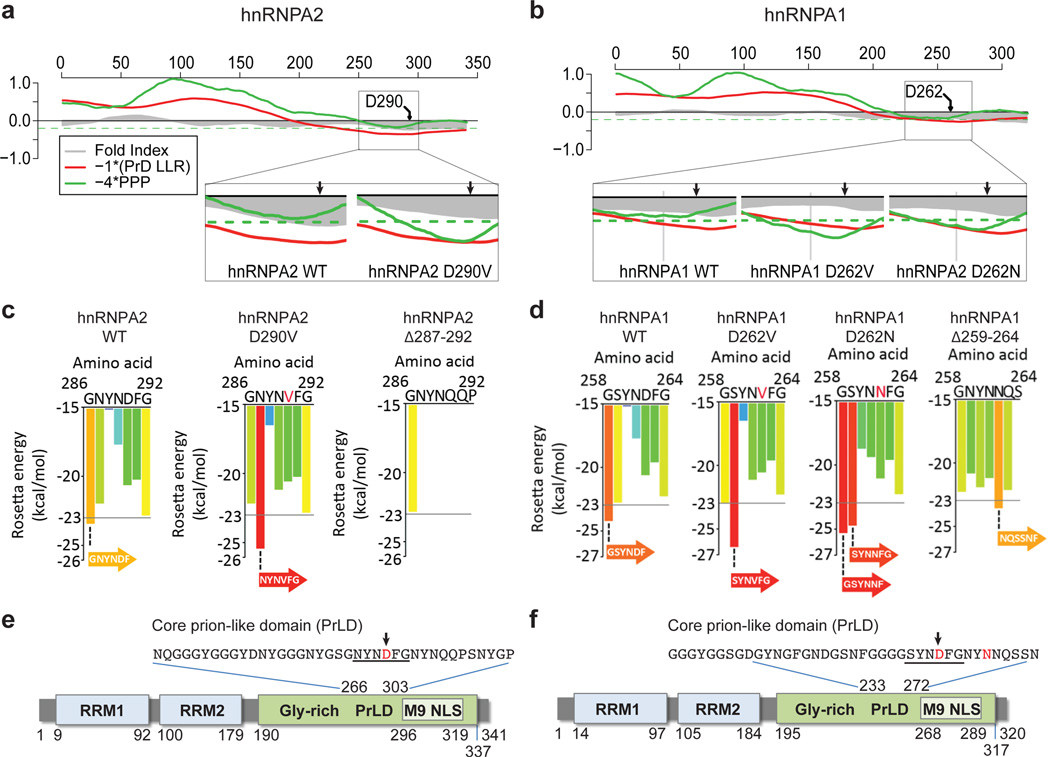
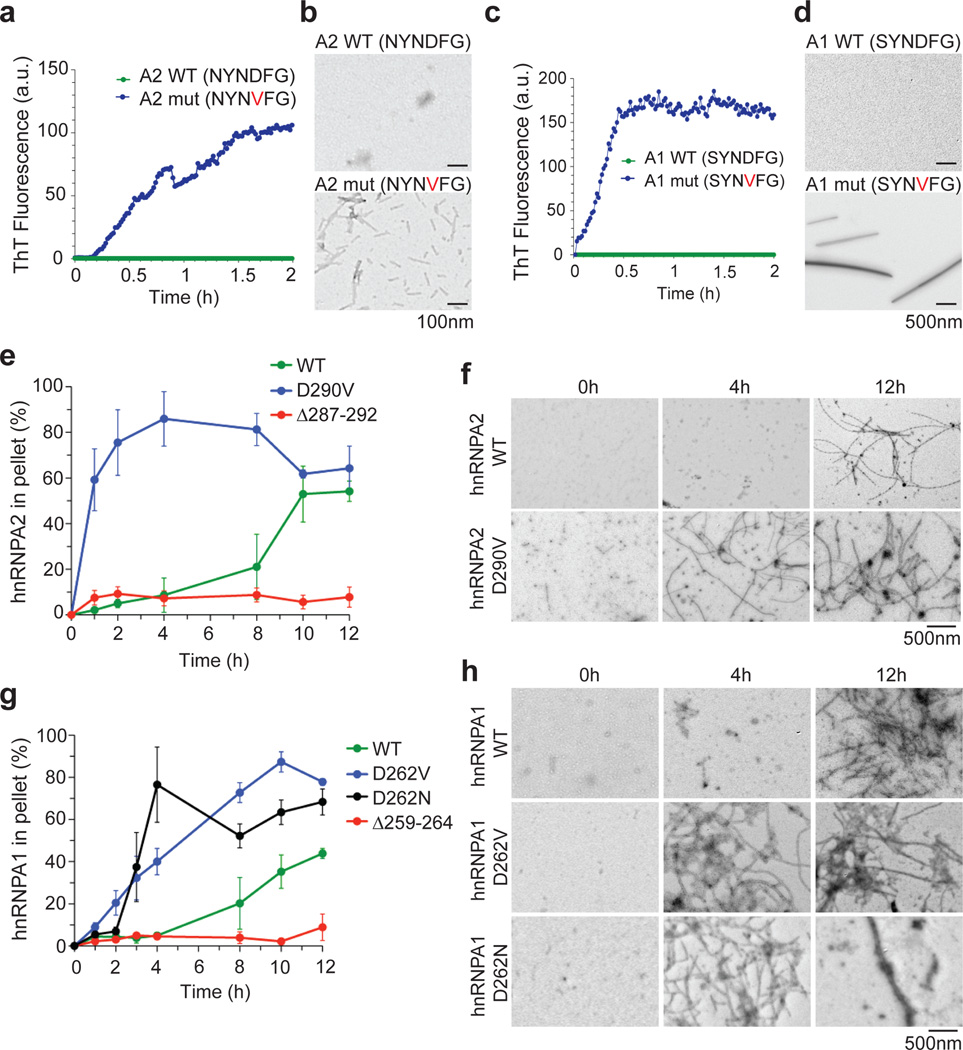
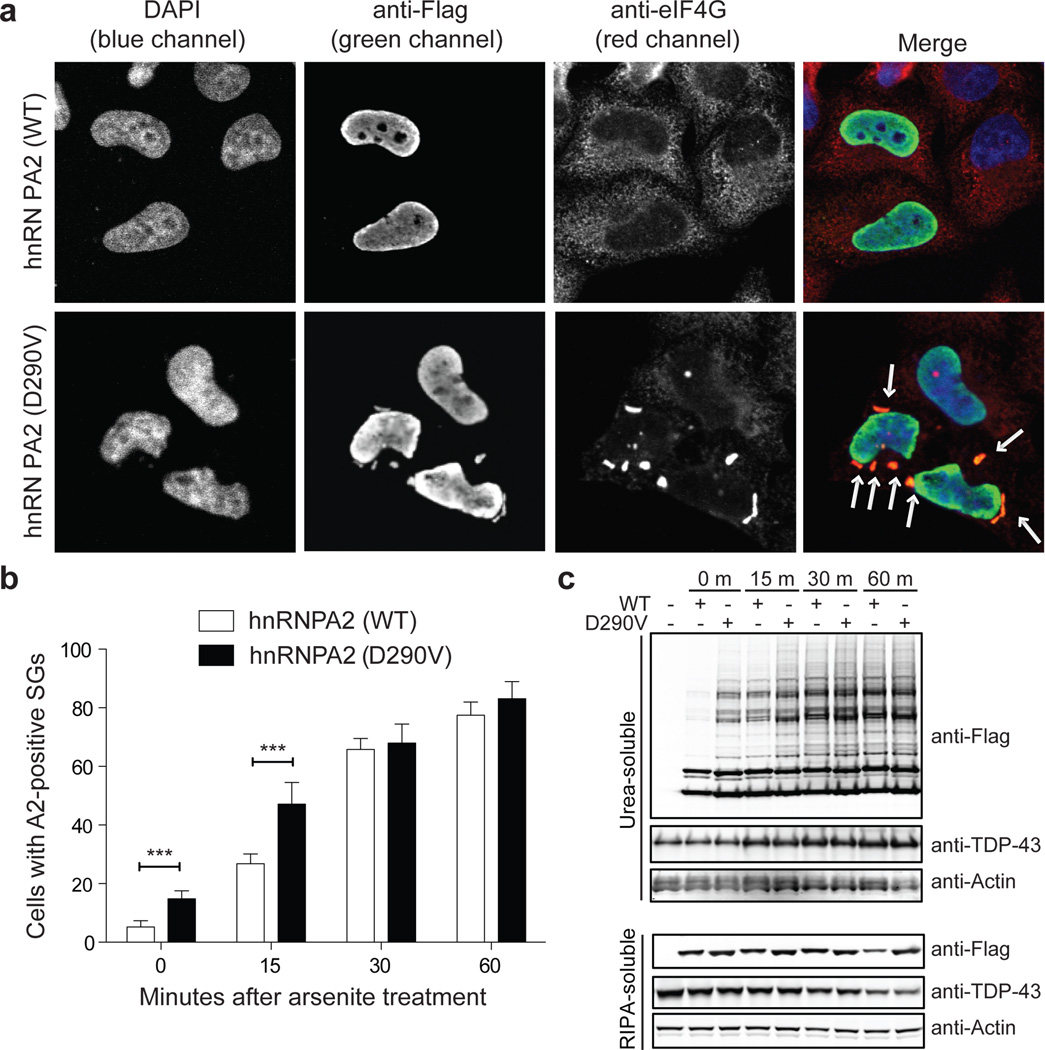
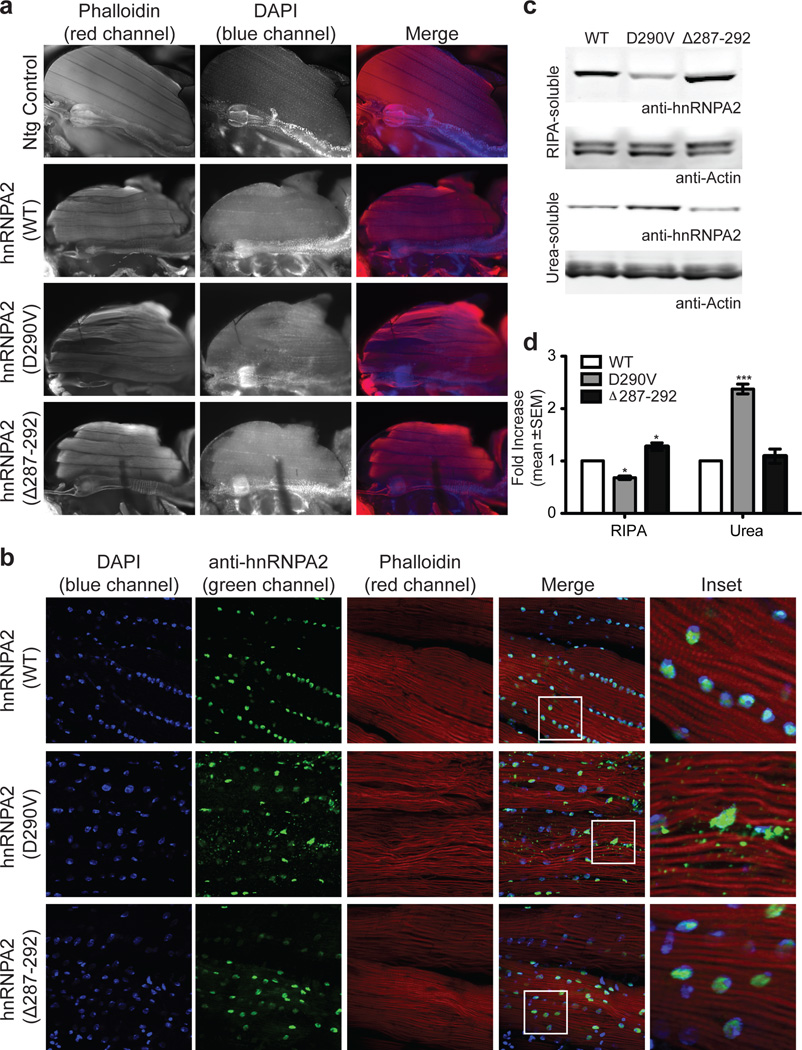
Algorithms designed to identify canonical yeast prions predict that around 250 human proteins, including several RNA-binding proteins associated with neurodegenerative disease, harbour a distinctive prion-like domain (PrLD) enriched in uncharged polar amino acids and glycine. PrLDs in RNA-binding proteins are essential for the assembly of ribonucleoprotein granules. However, the interplay between human PrLD function and disease is not understood. Here we define pathogenic mutations in PrLDs of heterogeneous nuclear ribonucleoproteins (hnRNPs) A2B1 and A1 in families with inherited degeneration affecting muscle, brain, motor neuron and bone, and in one case of familial amyotrophic lateral sclerosis. Wild-type hnRNPA2 (the most abundant isoform of hnRNPA2B1) and hnRNPA1 show an intrinsic tendency to assemble into self-seeding fibrils, which is exacerbated by the disease mutations. Indeed, the pathogenic mutations strengthen a 'steric zipper' motif in the PrLD, which accelerates the formation of self-seeding fibrils that cross-seed polymerization of wild-type hnRNP. Notably, the disease mutations promote excess incorporation of hnRNPA2 and hnRNPA1 into stress granules and drive the formation of cytoplasmic inclusions in animal models that recapitulate the human pathology. Thus, dysregulated polymerization caused by a potent mutant steric zipper motif in a PrLD can initiate degenerative disease. Related proteins with PrLDs should therefore be considered candidates for initiating and perhaps propagating proteinopathies of muscle, brain, motor neuron and bone.
Figures
References
-
- Nalbandian A, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci. 2011;45:522–531. - PubMed
-
- Watts GD, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. - PubMed
-
- Neumann M, Tolnay M, Mackenzie IR. The molecular basis of frontotemporal dementia. Expert Rev Mol Med. 2009;11:e23. - PubMed
-
- Shi Z, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol. 2011 - PubMed
References for Methods
-
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. - PubMed
-
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537:39–64. - PubMed
-
- Goode MG, Rodrigo AG. SQUINT: a multiple alignment program and editor. Bioinformatics. 2007;23:1553–1555. - PubMed
-
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph. D. dissertation, The University of Texas at Austin. 2006
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 089701/WT_/Wellcome Trust/United Kingdom
- MC_G1000733/MRC_/Medical Research Council/United Kingdom
- K02 AG042095/AG/NIA NIH HHS/United States
- R01 NS053825/NS/NINDS NIH HHS/United States
- AG031867/AG/NIA NIH HHS/United States
- G0900688/MRC_/Medical Research Council/United Kingdom
- R21 NS067354/NS/NINDS NIH HHS/United States
- P01 AG032953/AG/NIA NIH HHS/United States
- NS053825/NS/NINDS NIH HHS/United States
- DP2 OD002177/OD/NIH HHS/United States
- AG032953/AG/NIA NIH HHS/United States
- DP2OD002177/OD/NIH HHS/United States
- R01 AG031867/AG/NIA NIH HHS/United States
- NS067354/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
