

Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis
- PMID: 23457166
- PMCID: PMC9487424
- DOI: 10.1183/09059180.00008412
Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis
Abstract
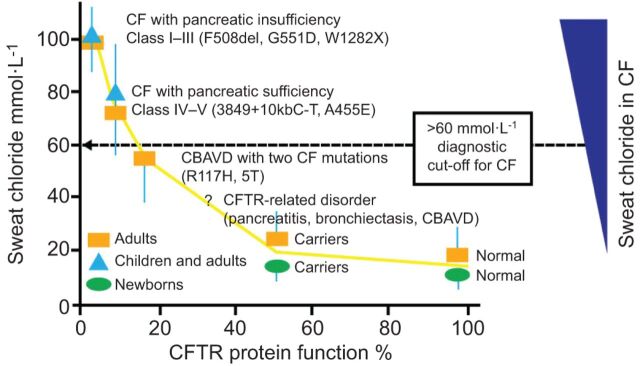
Cystic fibrosis (CF) is caused by genetic mutations that affect the cystic fibrosis transmembrane conductance regulator (CFTR) protein. These mutations can impact the synthesis and transfer of the CFTR protein to the apical membrane of epithelial cells, as well as influencing the gating or conductance of chloride and bicarbonate ions through the channel. CFTR dysfunction results in ionic imbalance of epithelial secretions in several organ systems, such as the pancreas, gastrointestinal tract, liver and the respiratory system. Since discovery of the CFTR gene in 1989, research has focussed on targeting the underlying genetic defect to identify a disease-modifying treatment for CF. Investigated management strategies have included gene therapy and the development of small molecules that target CFTR mutations, known as CFTR modulators. CFTR modulators are typically identified by high-throughput screening assays, followed by preclinical validation using cell culture systems. Recently, one such modulator, the CFTR potentiator ivacaftor, was approved as an oral therapy for CF patients with the G551D-CFTR mutation. The clinical development of ivacaftor not only represents a breakthrough in CF care but also serves as a noteworthy example of personalised medicine.
Conflict of interest statement
N. Derichs received speaker honorarium from Vertex Pharmaceuticals Inc. for participation in a symposium and served as a consultant for Vertex Inc. at educational activities and advisory boards.
Figures


References
-
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Childhood 1938; 56: 344–399.
-
- Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996; 154: 1229–1256. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, et al. . Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073–1080. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, et al. . Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–1073. - PubMed
-
- Rommens JM, Iannuzzi MC, Kerem B, et al. . Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245: 1059–1065. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical