

Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation
- PMID: 23457167
- PMCID: PMC9487423
- DOI: 10.1183/09059180.00008512
Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation
Abstract
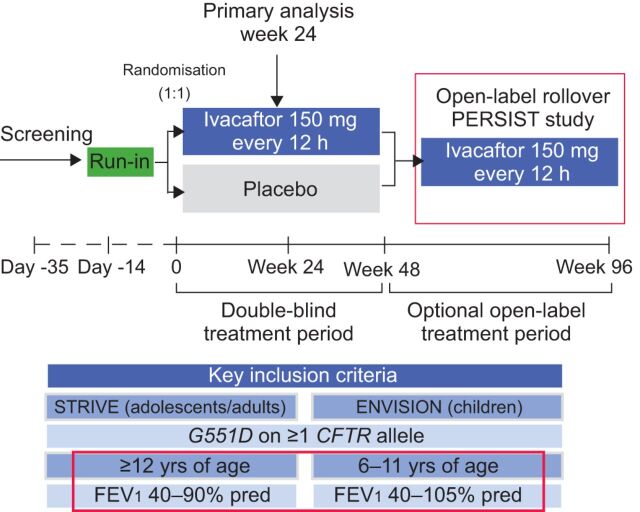
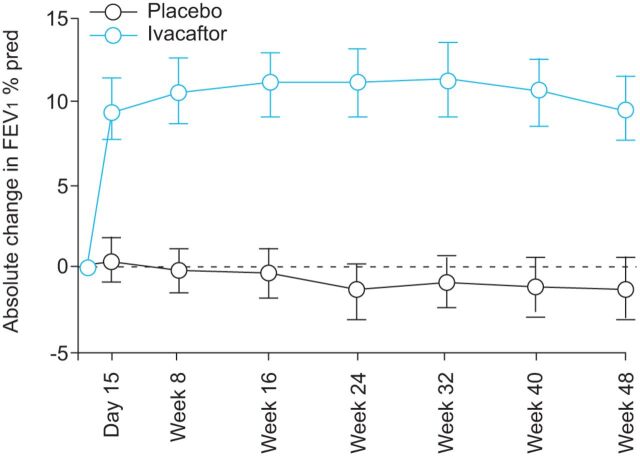
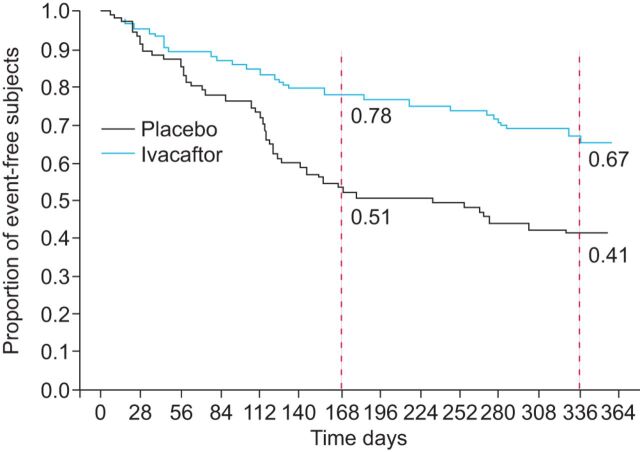
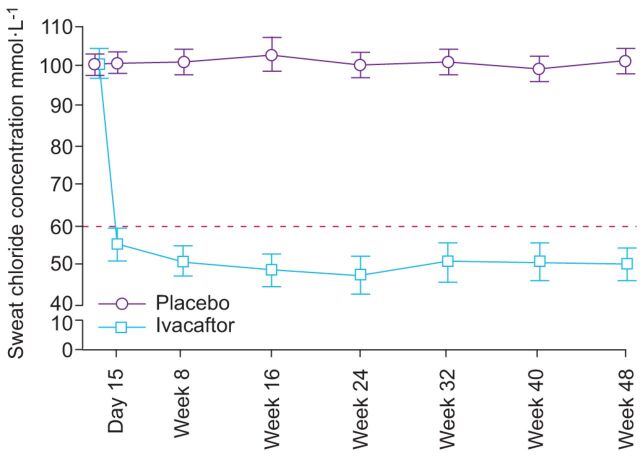
Cystic fibrosis (CF) is an autosomal recessive lethal disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes for CFTR, an epithelial cell-surface expressed protein responsible for the transport of chloride (Cl(-)). Gating mutations associated with defective conductance can be modulated by CFTR potentiators. Ivacaftor is a CFTR potentiator approved for the treatment of CF patients >6 yrs of age with at least one copy of the G551D-CFTR mutation. Herein, the clinical trial development programme for ivacaftor will be reviewed, including two pivotal studies in adolescents/adults and in children. These studies report sustained improvements in lung function and sweat chloride concentrations, and a reduction in pulmonary exacerbations over a 48-week treatment period. In the era of personalised medicine, ivacaftor offers an effective and well-tolerated treatment for the clinical management of CF patients with the G551D mutation. A long-term, open-label study will report the effects of ivacaftor over a further 48 weeks.
Conflict of interest statement
I. Sermet-Gaudelus has received speaker honorarium and reimbursement for attending a symposium from Vertex Inc.
Figures
References
-
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992–2001. - PubMed
-
- Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011; 139: 1480–1490. - PubMed
-
- Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 2004; 23: 146–158. - PubMed
-
- De Boeck K, Kent L, Davies J, et al. . CFTR biomarkers: time for promotion to surrogate end-point. Eur Respir J 2013; 41: 203–216. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical