

Dysregulation of multiple facets of glycogen metabolism in a murine model of Pompe disease
- PMID: 23457523
- PMCID: PMC3572993
- DOI: 10.1371/journal.pone.0056181
Dysregulation of multiple facets of glycogen metabolism in a murine model of Pompe disease
Abstract
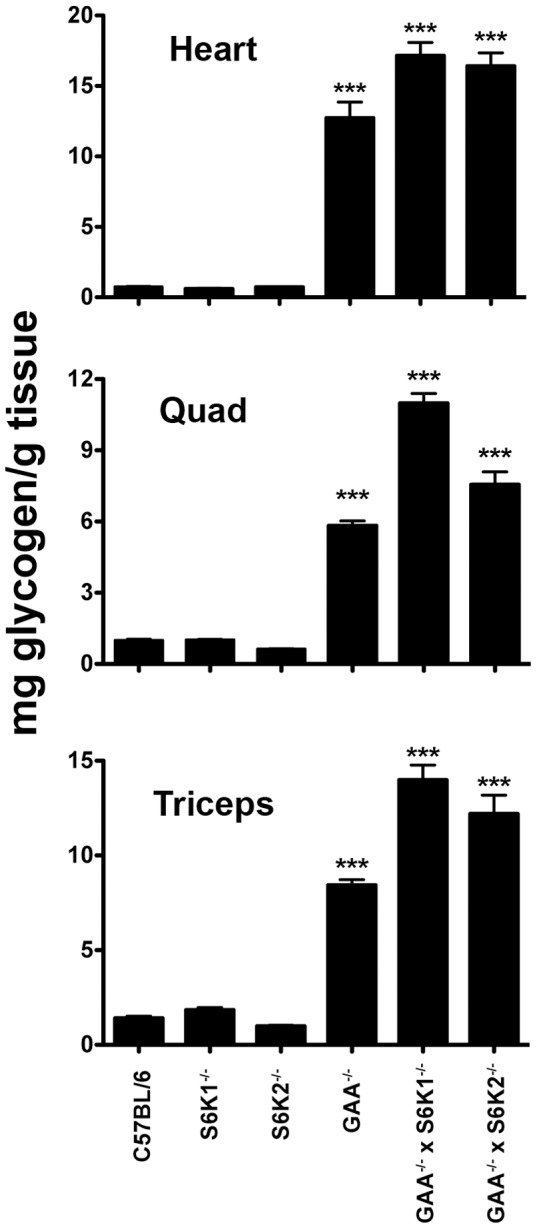
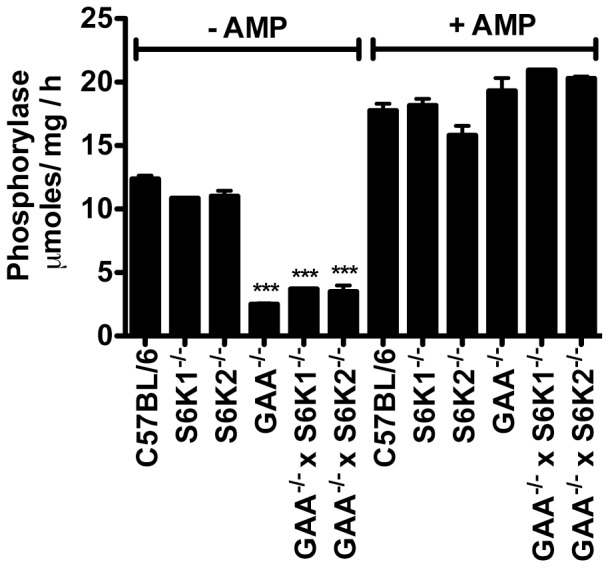
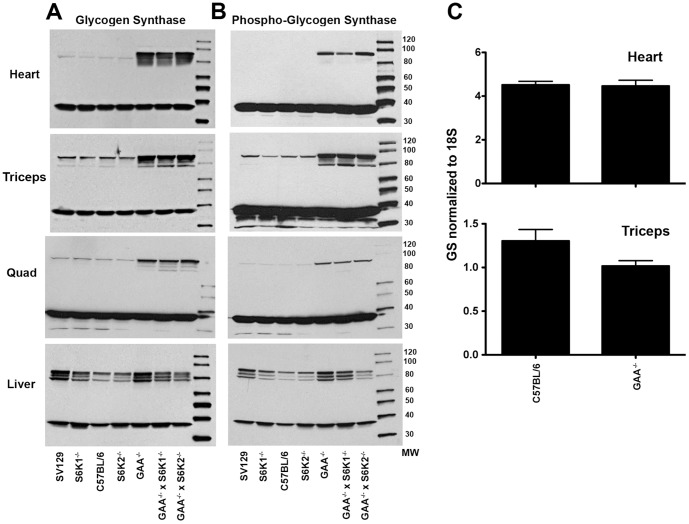
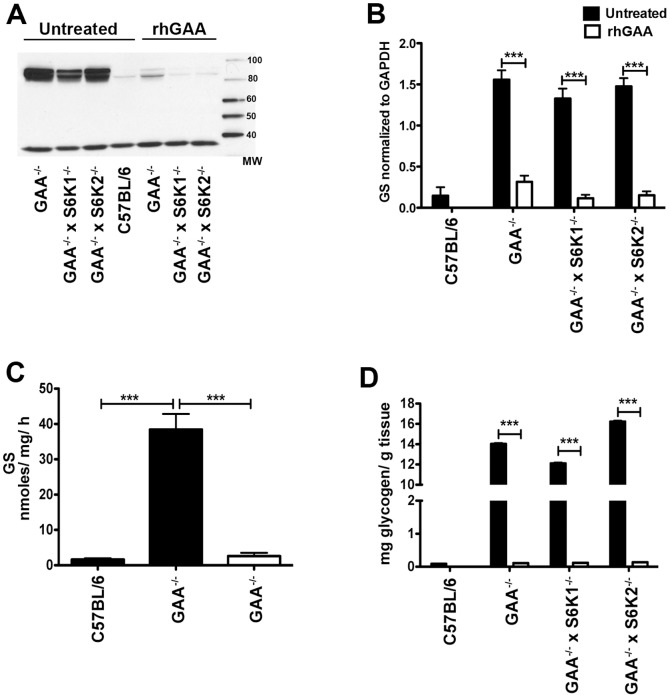
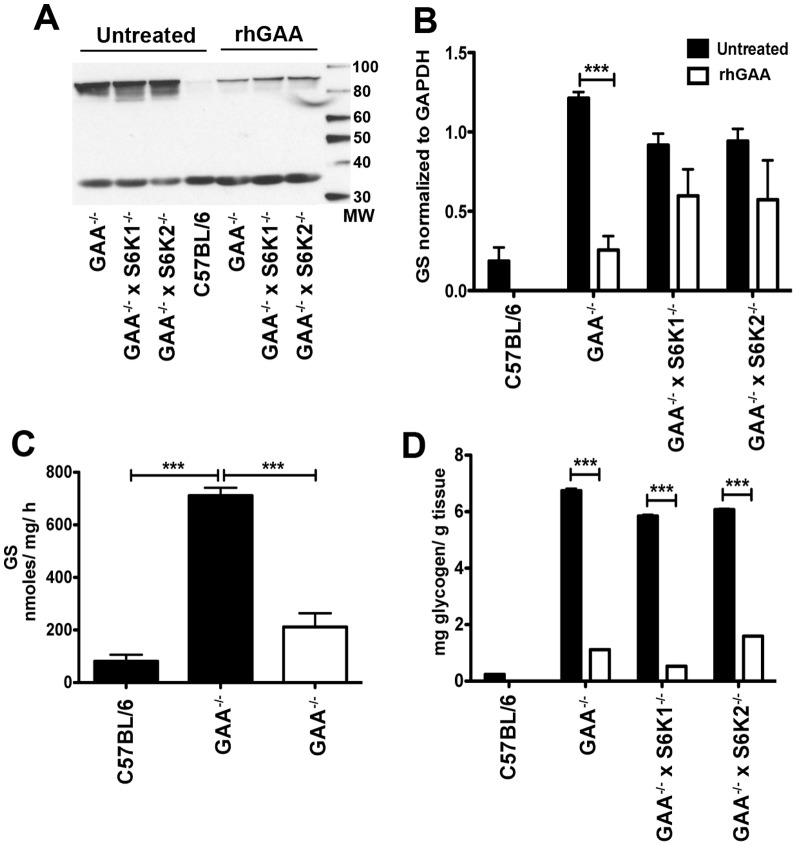
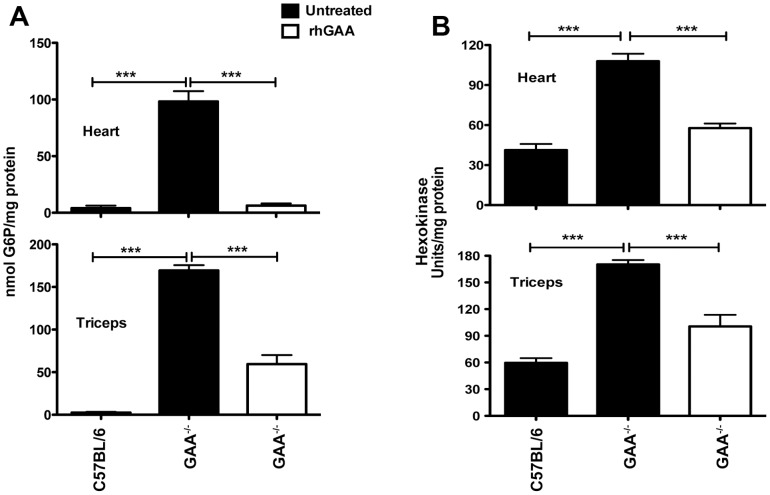
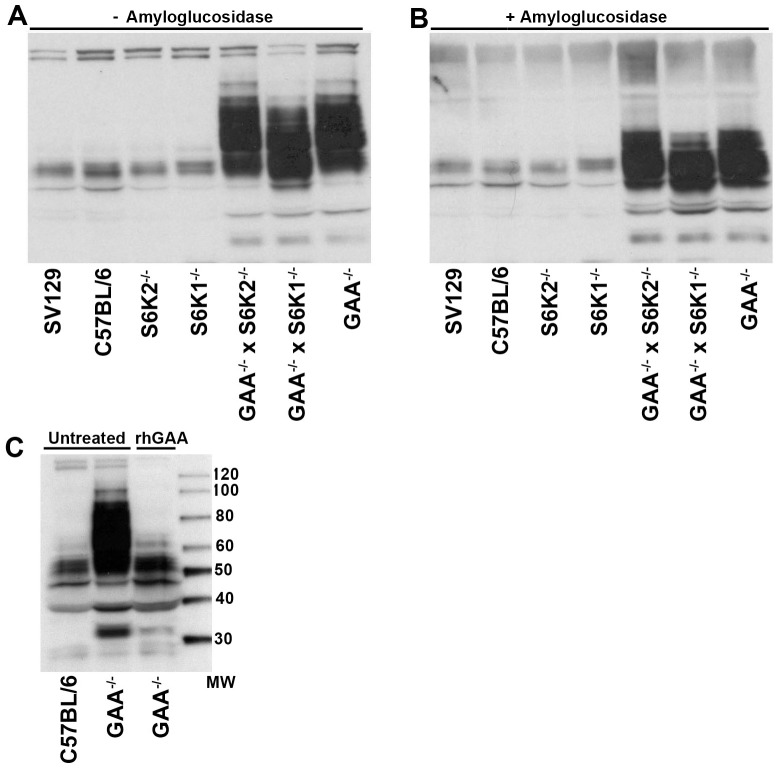
Pompe disease, also known as glycogen storage disease (GSD) type II, is caused by deficiency of lysosomal acid α-glucosidase (GAA). The resulting glycogen accumulation causes a spectrum of disease severity ranging from a rapidly progressive course that is typically fatal by 1 to 2 years of age to a slower progressive course that causes significant morbidity and early mortality in children and adults. The aim of this study is to better understand the biochemical consequences of glycogen accumulation in the Pompe mouse. We evaluated glycogen metabolism in heart, triceps, quadriceps, and liver from wild type and several strains of GAA(-/-) mice. Unexpectedly, we observed that lysosomal glycogen storage correlated with a robust increase in factors that normally promote glycogen biosynthesis. The GAA(-/-) mouse strains were found to have elevated glycogen synthase (GS), glycogenin, hexokinase, and glucose-6-phosphate (G-6-P, the allosteric activator of GS). Treating GAA(-/-) mice with recombinant human GAA (rhGAA) led to a dramatic reduction in the levels of glycogen, GS, glycogenin, and G-6-P. Lysosomal glycogen storage also correlated with a dysregulation of phosphorylase, which normally breaks down cytoplasmic glycogen. Analysis of phosphorylase activity confirmed a previous report that, although phosphorylase protein levels are identical in muscle lysates from wild type and GAA(-/-) mice, phosphorylase activity is suppressed in the GAA(-/-) mice in the absence of AMP. This reduction in phosphorylase activity likely exacerbates lysosomal glycogen accumulation. If the dysregulation in glycogen metabolism observed in the mouse model of Pompe disease also occurs in Pompe patients, it may contribute to the observed broad spectrum of disease severity.
Conflict of interest statement
Figures
References
-
- Engel AG, Hirschhorn R, Huie ML (2004) Acid Maltase Deficiency. Myology. 3rd ed. New York: McGraw-Hill. pp. 1559–1586.
-
- Kroos MA, Pomponio RJ, Hagemans ML, Keulemans JL, Phipps M, et al. (2007) Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 68: 110–115. - PubMed
-
- Orth M, Mundegar RR (2003) Effect of acid maltase deficiency on the endosomal/lysosomal system and glucose transporter 4. Neuromuscul Disord 13: 49–54. - PubMed
-
- Ashe KM, Taylor KM, Chu Q, Meyers E, Ellis A, et al. (2010) Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Molecular Genetics and Metabolism 100: 309–315. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
