

Canonical correlation analysis for RNA-seq co-expression networks
- PMID: 23460206
- PMCID: PMC3632131
- DOI: 10.1093/nar/gkt145
Canonical correlation analysis for RNA-seq co-expression networks
Abstract
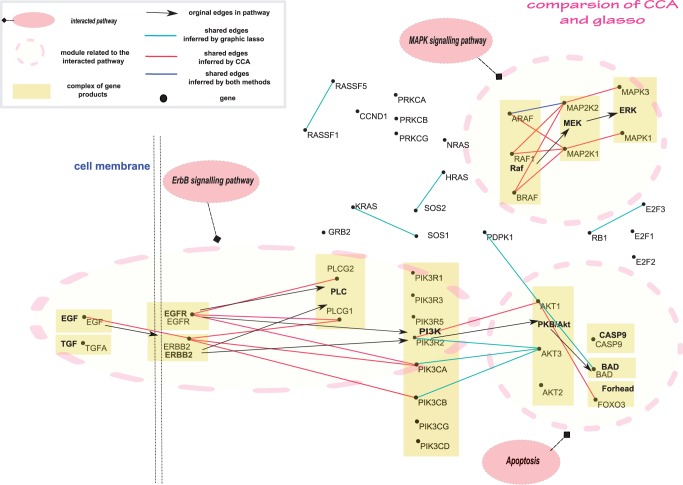
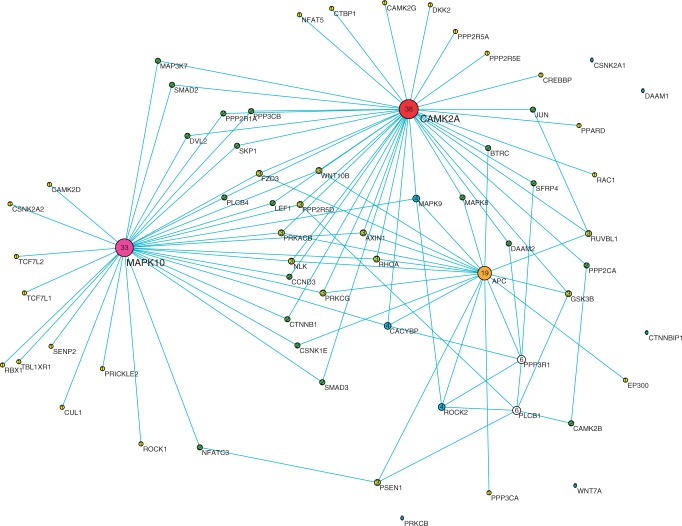
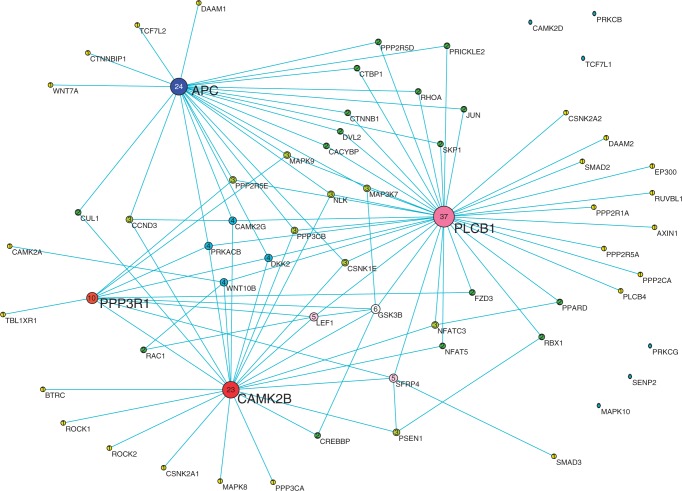
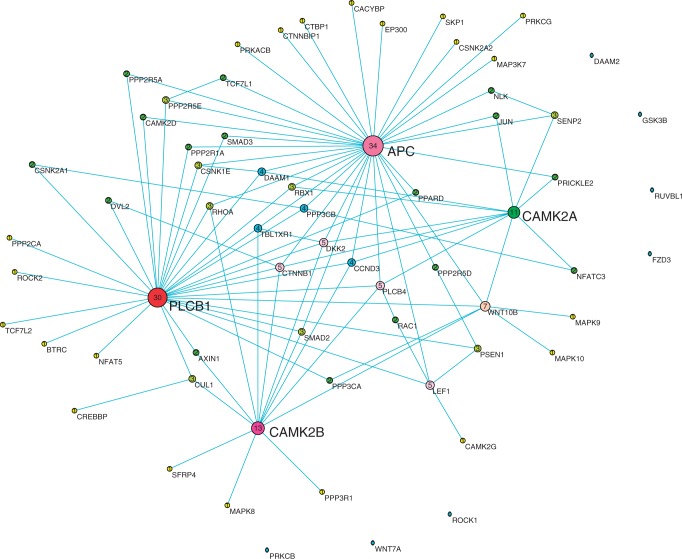
Digital transcriptome analysis by next-generation sequencing discovers substantial mRNA variants. Variation in gene expression underlies many biological processes and holds a key to unravelling mechanism of common diseases. However, the current methods for construction of co-expression networks using overall gene expression are originally designed for microarray expression data, and they overlook a large number of variations in gene expressions. To use information on exon, genomic positional level and allele-specific expressions, we develop novel component-based methods, single and bivariate canonical correlation analysis, for construction of co-expression networks with RNA-seq data. To evaluate the performance of our methods for co-expression network inference with RNA-seq data, they are applied to lung squamous cell cancer expression data from TCGA database and our bipolar disorder and schizophrenia RNA-seq study. The preliminary results demonstrate that the co-expression networks constructed by canonical correlation analysis and RNA-seq data provide rich genetic and molecular information to gain insight into biological processes and disease mechanism. Our new methods substantially outperform the current statistical methods for co-expression network construction with microarray expression data or RNA-seq data based on overall gene expression levels.
Figures
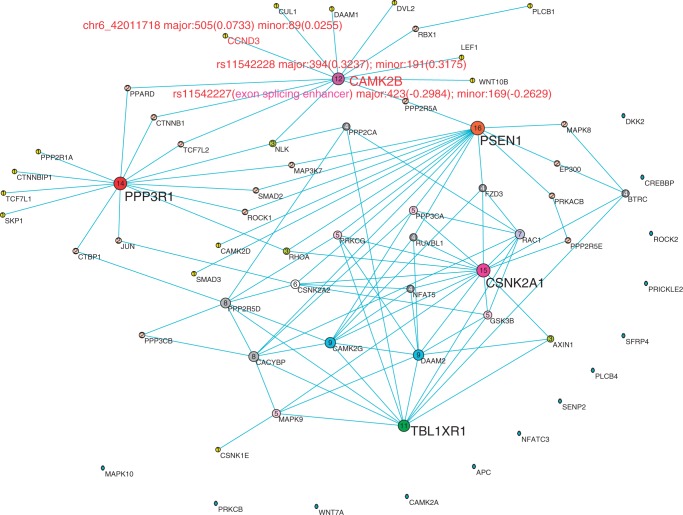
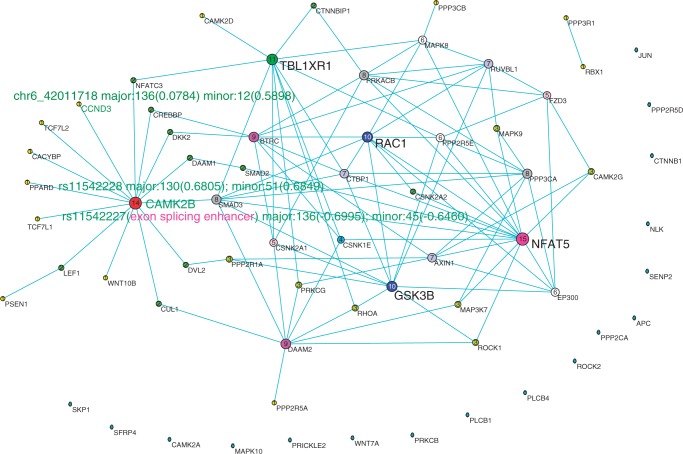
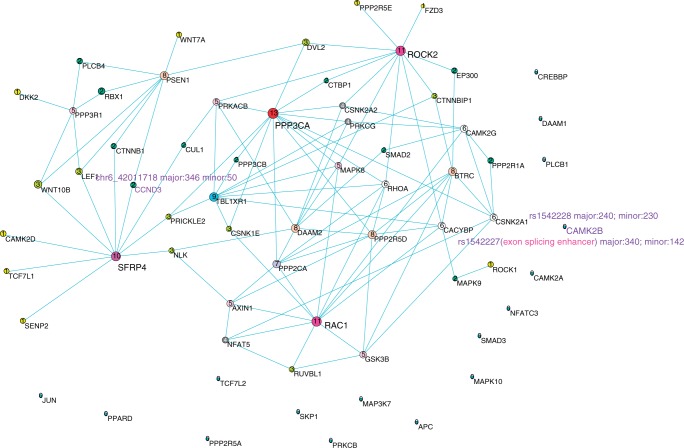
References
-
- Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012;10:618–630. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
