

Entering the second century of maize quantitative genetics
- PMID: 23462502
- PMCID: PMC3860165
- DOI: 10.1038/hdy.2013.6
Entering the second century of maize quantitative genetics
Abstract
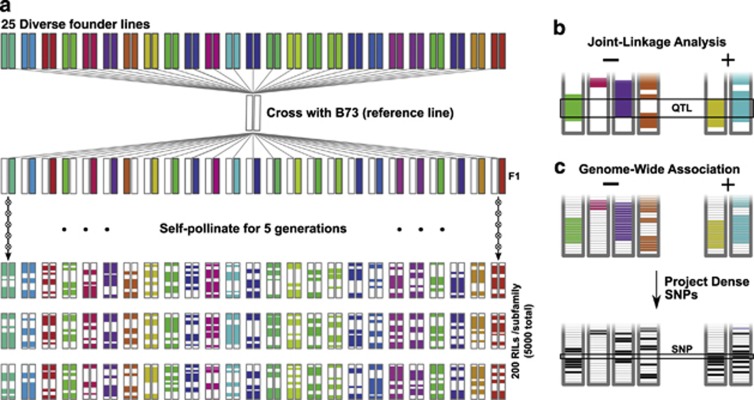
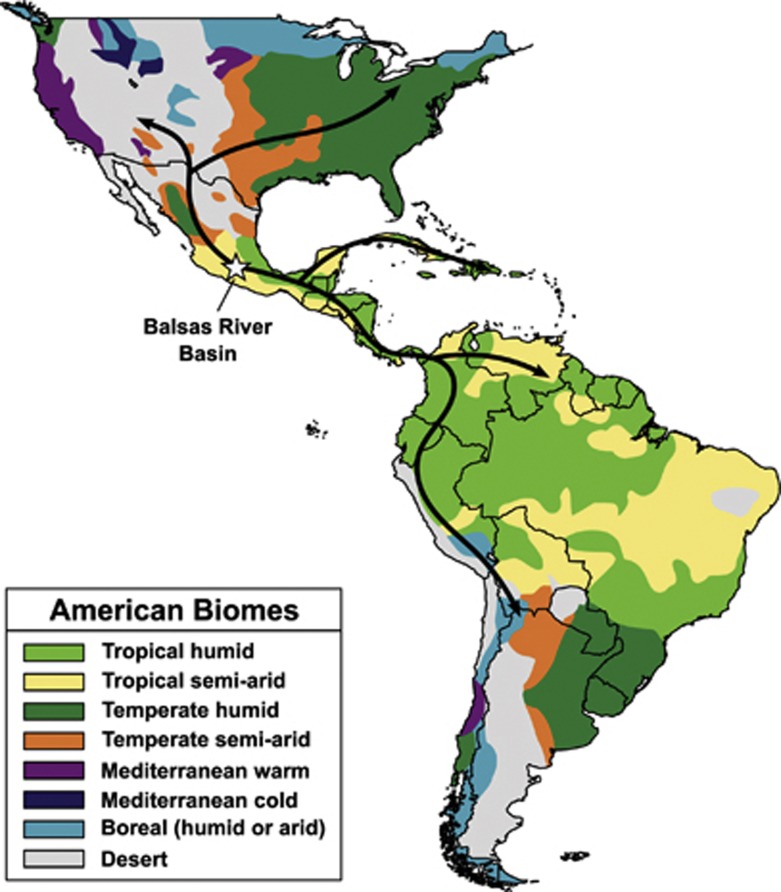
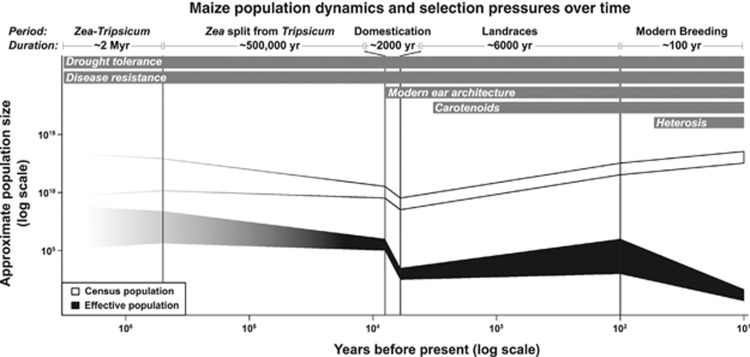
Maize is the most widely grown cereal in the world. In addition to its role in global agriculture, it has also long served as a model organism for genetic research. Maize stands at a genetic crossroads, as it has access to all the tools available for plant genetics but exhibits a genetic architecture more similar to other outcrossing organisms than to self-pollinating crops and model plants. In this review, we summarize recent advances in maize genetics, including the development of powerful populations for genetic mapping and genome-wide association studies (GWAS), and the insights these studies yield on the mechanisms underlying complex maize traits. Most maize traits are controlled by a large number of genes, and linkage analysis of several traits implicates a 'common gene, rare allele' model of genetic variation where some genes have many individually rare alleles contributing. Most natural alleles exhibit small effect sizes with little-to-no detectable pleiotropy or epistasis. Additionally, many of these genes are locked away in low-recombination regions that encourage the formation of multi-gene blocks that may underlie maize's strong heterotic effect. Domestication left strong marks on the maize genome, and some of the differences in trait architectures may be due to different selective pressures over time. Overall, maize's advantages as a model system make it highly desirable for studying the genetics of outcrossing species, and results from it can provide insight into other such species, including humans.
Figures
Similar articles
-
Genetic Architecture of Domestication-Related Traits in Maize.Genetics. 2016 Sep;204(1):99-113. doi: 10.1534/genetics.116.191106. Epub 2016 Jul 13. Genetics. 2016. PMID: 27412713 Free PMC article.
-
Genome-wide association study of leaf architecture in the maize nested association mapping population.Nat Genet. 2011 Feb;43(2):159-62. doi: 10.1038/ng.746. Epub 2011 Jan 9. Nat Genet. 2011. PMID: 21217756
-
Heterotic trait locus (HTL) mapping identifies intra-locus interactions that underlie reproductive hybrid vigor in Sorghum bicolor.PLoS One. 2012;7(6):e38993. doi: 10.1371/journal.pone.0038993. Epub 2012 Jun 25. PLoS One. 2012. PMID: 22761720 Free PMC article.
-
Tracking footprints of maize domestication and evidence for a massive selective sweep on chromosome 10.Proc Natl Acad Sci U S A. 2009 Jun 16;106 Suppl 1(Suppl 1):9979-86. doi: 10.1073/pnas.0901122106. Epub 2009 Jun 15. Proc Natl Acad Sci U S A. 2009. PMID: 19528660 Free PMC article. Review.
-
Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize.Int J Mol Sci. 2024 Feb 5;25(3):1918. doi: 10.3390/ijms25031918. Int J Mol Sci. 2024. PMID: 38339196 Free PMC article. Review.
Cited by
-
Modelling the genetic architecture of flowering time control in barley through nested association mapping.BMC Genomics. 2015 Apr 12;16(1):290. doi: 10.1186/s12864-015-1459-7. BMC Genomics. 2015. PMID: 25887319 Free PMC article.
-
Back to the wild: mining maize (Zea mays L.) disease resistance using advanced breeding tools.Mol Biol Rep. 2022 Jun;49(6):5787-5803. doi: 10.1007/s11033-021-06815-x. Epub 2022 Jan 22. Mol Biol Rep. 2022. PMID: 35064401
-
Genetic analysis of safflower domestication.BMC Plant Biol. 2014 Feb 6;14:43. doi: 10.1186/1471-2229-14-43. BMC Plant Biol. 2014. PMID: 24502326 Free PMC article.
-
The genetic architecture of leaf stable carbon isotope composition in Zea mays and the effect of transpiration efficiency on leaf elemental accumulation.G3 (Bethesda). 2021 Sep 6;11(9):jkab222. doi: 10.1093/g3journal/jkab222. G3 (Bethesda). 2021. PMID: 34544133 Free PMC article.
-
Mapping of QTLs for morphophysiological and yield traits under water-deficit stress and well-watered conditions in maize.Front Plant Sci. 2023 May 8;14:1124619. doi: 10.3389/fpls.2023.1124619. eCollection 2023. Front Plant Sci. 2023. PMID: 37223807 Free PMC article.
References
-
- Albrecht T, Wimmer V, Auinger H-J, Erbe M, Knaak C, Ouzunova M, et al. Genome-based prediction of testcross values in maize. Theor Appl Genet. 2011;123:339–350. - PubMed
-
- Beadle G. The ancestry of corn. Scientific American. 1980;242:112–119.
-
- Beló A, Zheng P, Luck S, Shen B, Meyer DJ, Li B, et al. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol Genet Genomics. 2008;279:1–10. - PubMed
-
- Bernardo R. Genomewide selection for rapid introgression of exotic germplasm in maize. Crop Sci. 2009;49:419–425.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
