

Patterns of population epigenomic diversity
- PMID: 23467092
- PMCID: PMC3798000
- DOI: 10.1038/nature11968
Patterns of population epigenomic diversity
Abstract
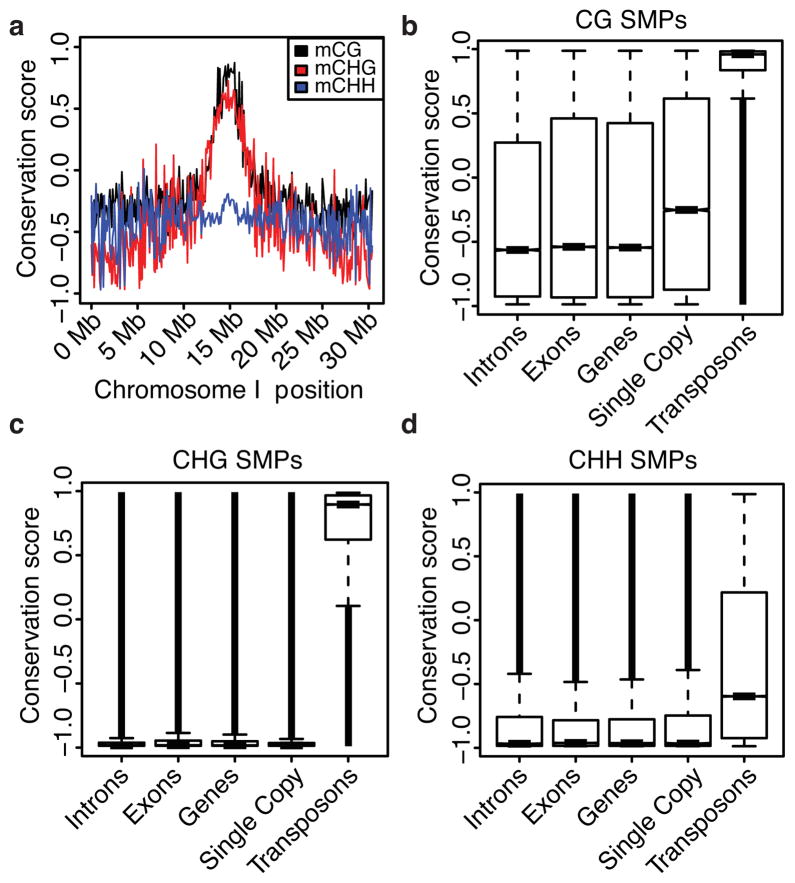
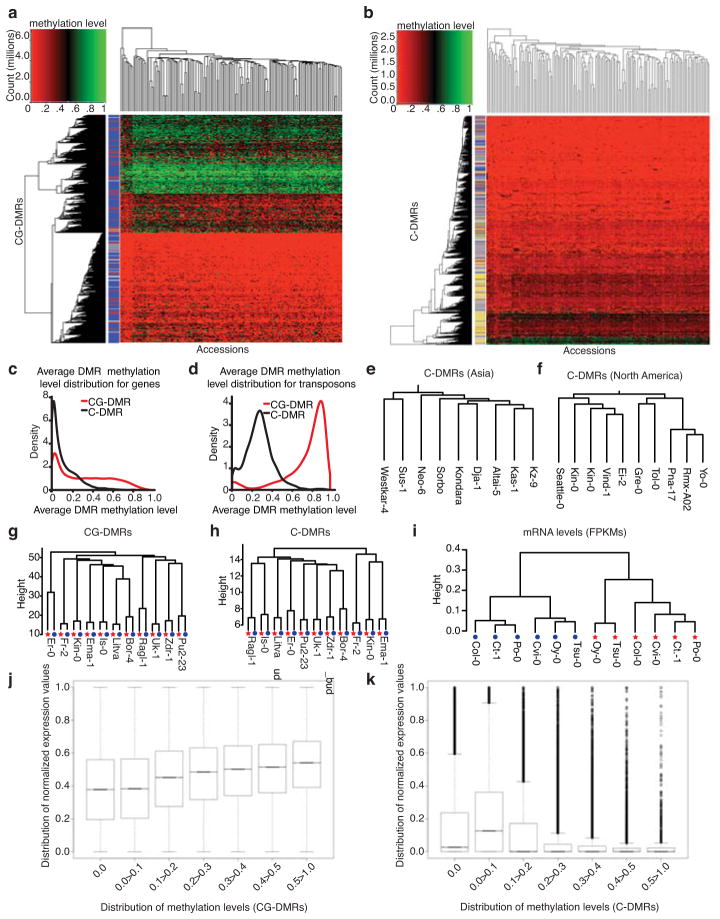
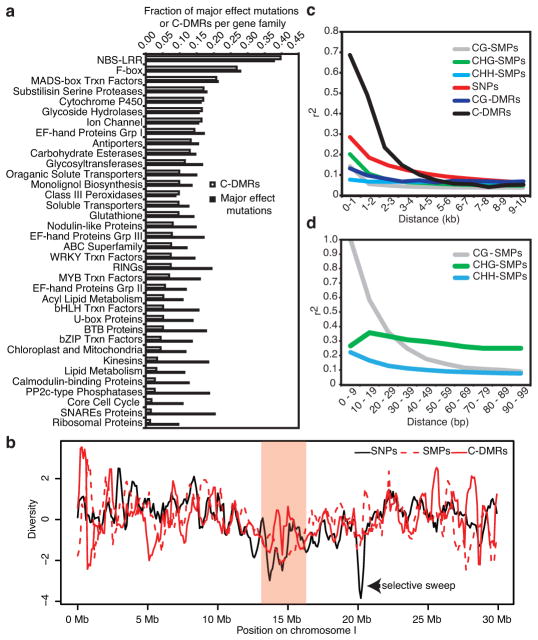
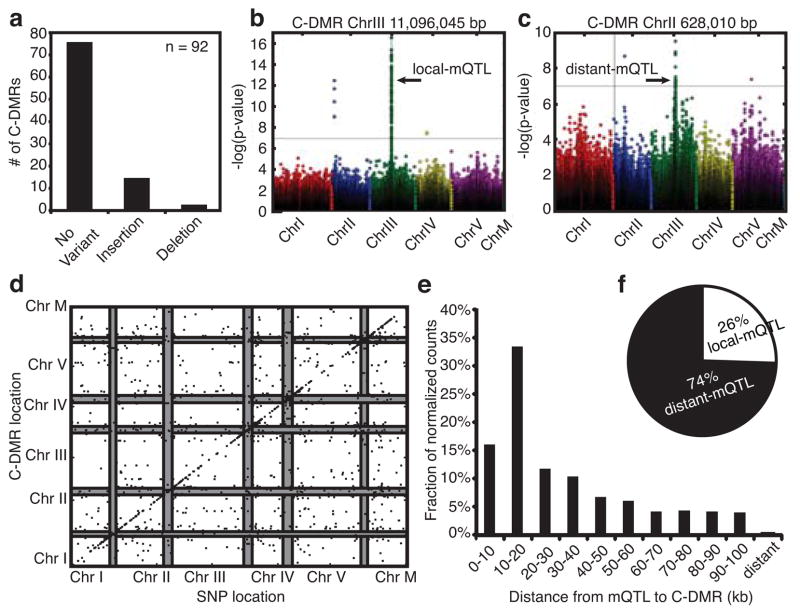
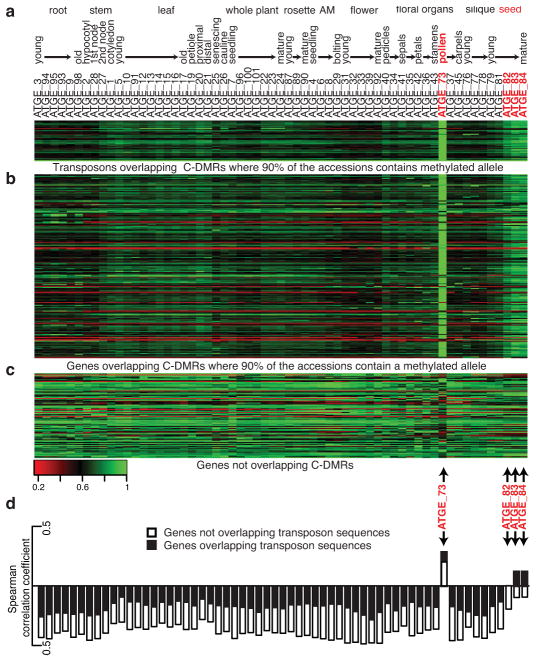
Natural epigenetic variation provides a source for the generation of phenotypic diversity, but to understand its contribution to such diversity, its interaction with genetic variation requires further investigation. Here we report population-wide DNA sequencing of genomes, transcriptomes and methylomes of wild Arabidopsis thaliana accessions. Single cytosine methylation polymorphisms are not linked to genotype. However, the rate of linkage disequilibrium decay amongst differentially methylated regions targeted by RNA-directed DNA methylation is similar to the rate for single nucleotide polymorphisms. Association analyses of these RNA-directed DNA methylation regions with genetic variants identified thousands of methylation quantitative trait loci, which revealed the population estimate of genetically dependent methylation variation. Analysis of invariably methylated transposons and genes across this population indicates that loci targeted by RNA-directed DNA methylation are epigenetically activated in pollen and seeds, which facilitates proper development of these structures.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Epigenomics: Methylation's mark on inheritance.Nature. 2013 Mar 14;495(7440):181-2. doi: 10.1038/nature11960. Epub 2013 Mar 6. Nature. 2013. PMID: 23467093 No abstract available.
References
-
- Becker C, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–249. - PubMed
Methods References
-
- Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society. 2002;64:479–498.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
