

Deleterious alleles in the human genome are on average younger than neutral alleles of the same frequency
- PMID: 23468643
- PMCID: PMC3585140
- DOI: 10.1371/journal.pgen.1003301
Deleterious alleles in the human genome are on average younger than neutral alleles of the same frequency
Abstract
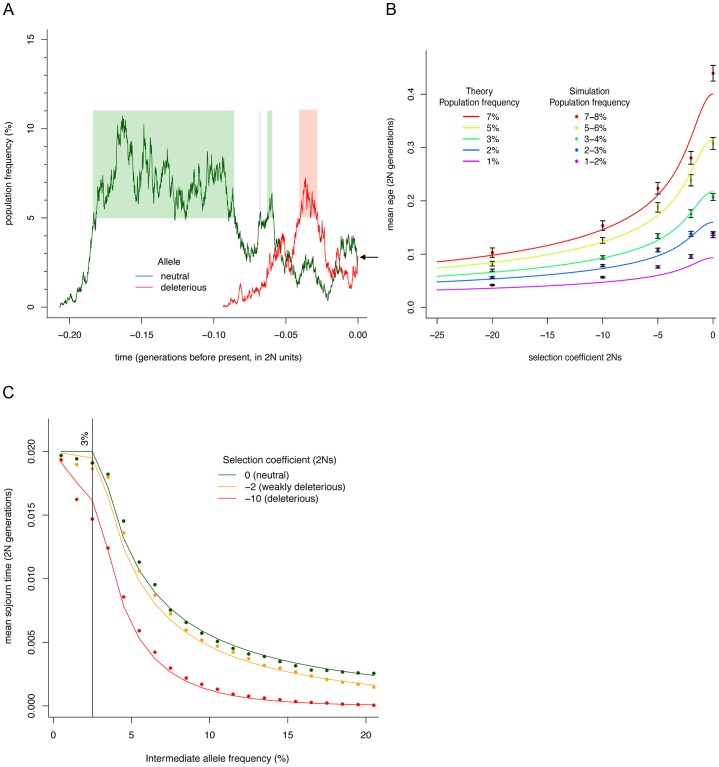
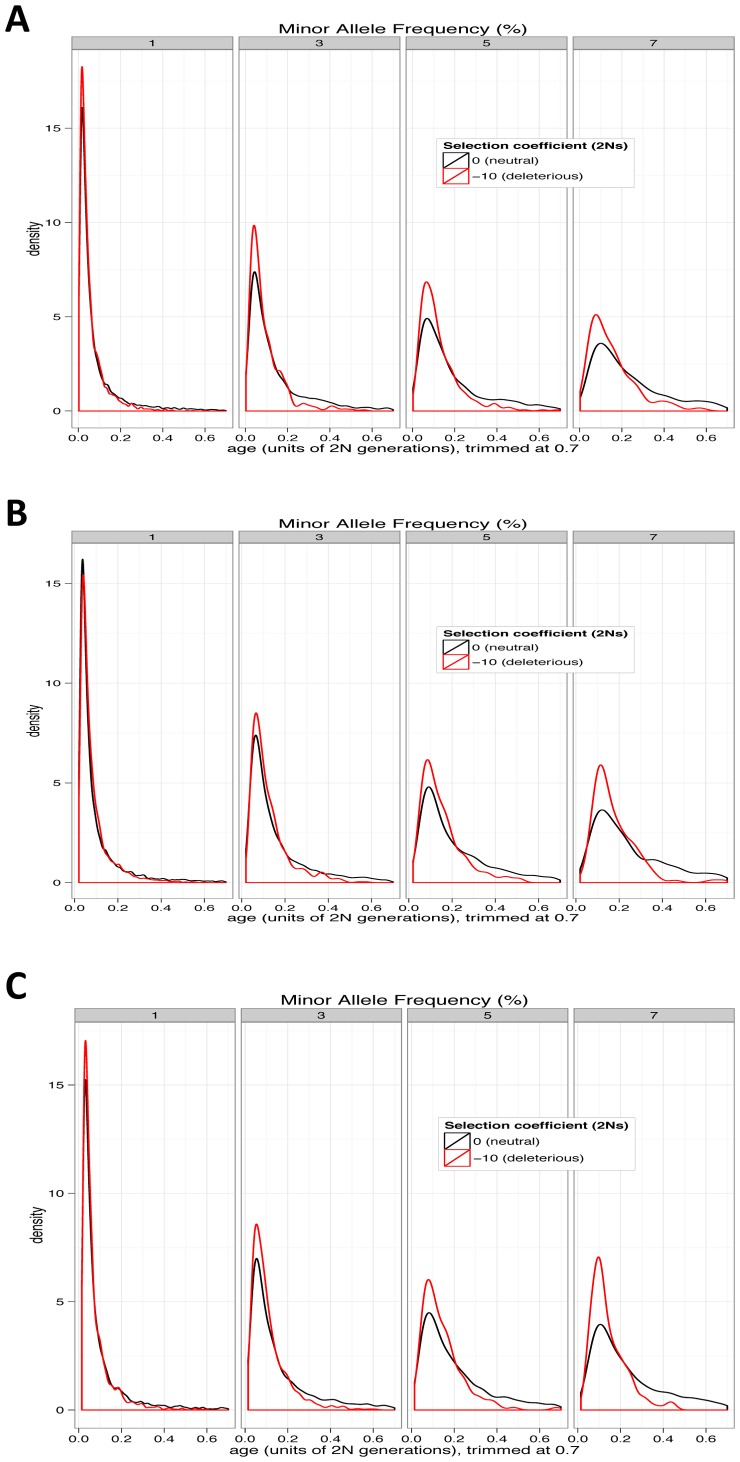
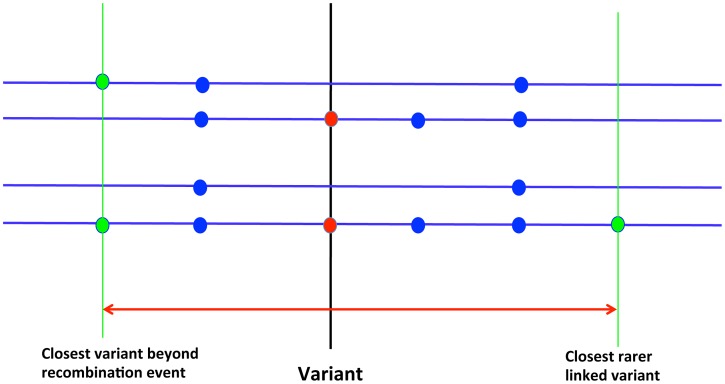
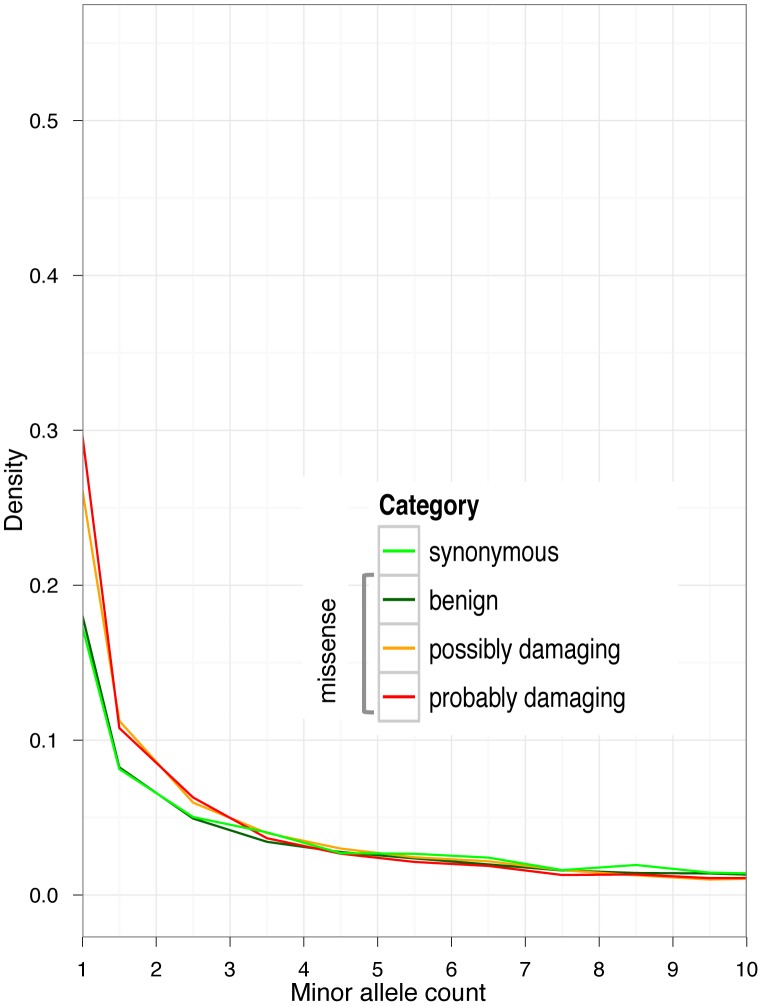
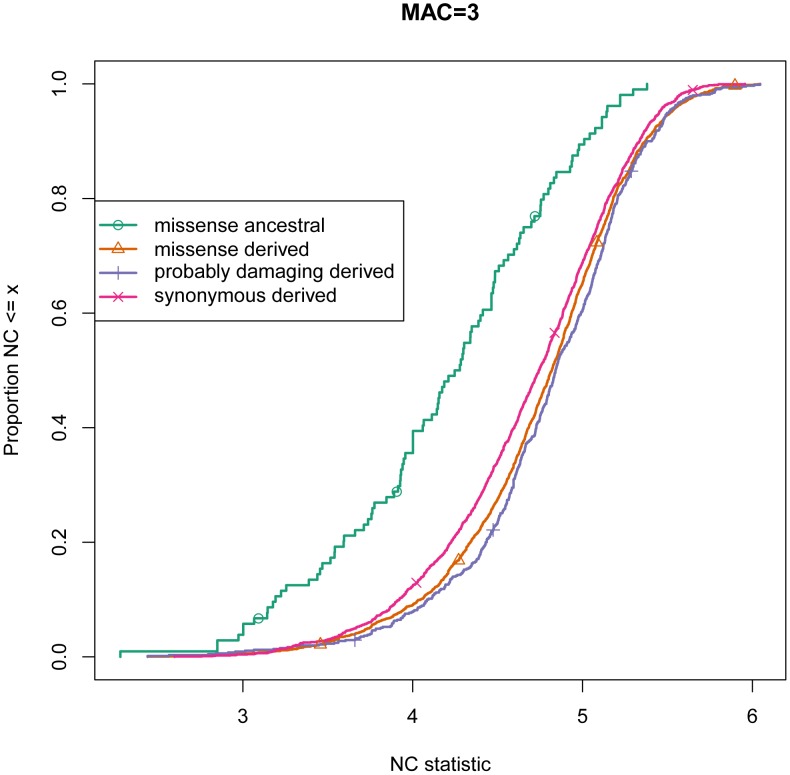
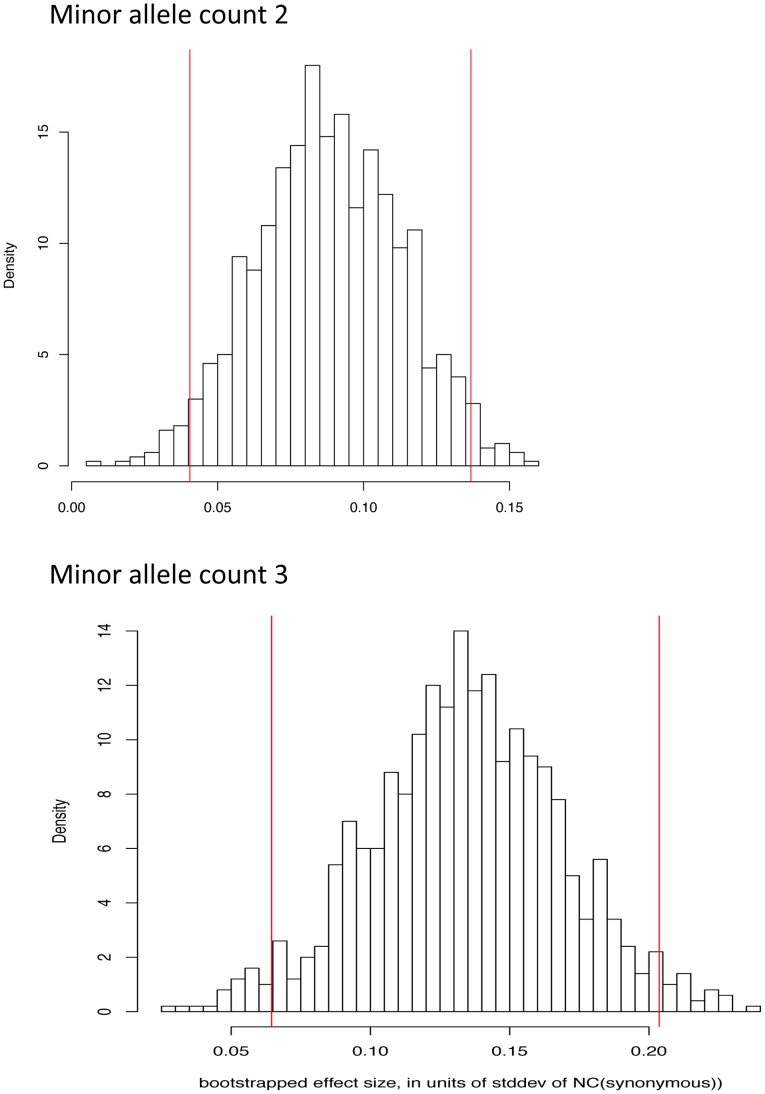
Large-scale population sequencing studies provide a complete picture of human genetic variation within the studied populations. A key challenge is to identify, among the myriad alleles, those variants that have an effect on molecular function, phenotypes, and reproductive fitness. Most non-neutral variation consists of deleterious alleles segregating at low population frequency due to incessant mutation. To date, studies characterizing selection against deleterious alleles have been based on allele frequency (testing for a relative excess of rare alleles) or ratio of polymorphism to divergence (testing for a relative increase in the number of polymorphic alleles). Here, starting from Maruyama's theoretical prediction (Maruyama T (1974), Am J Hum Genet USA 6:669-673) that a (slightly) deleterious allele is, on average, younger than a neutral allele segregating at the same frequency, we devised an approach to characterize selection based on allelic age. Unlike existing methods, it compares sets of neutral and deleterious sequence variants at the same allele frequency. When applied to human sequence data from the Genome of the Netherlands Project, our approach distinguishes low-frequency coding non-synonymous variants from synonymous and non-coding variants at the same allele frequency and discriminates between sets of variants independently predicted to be benign or damaging for protein structure and function. The results confirm the abundance of slightly deleterious coding variation in humans.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Sunyaev S, Ramensky V, Koch I, Lathe W, Kondrashov AS, et al. (2001) Prediction of deleterious human alleles. Human Molecular Genetics 10: 591–597. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
