

Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury
- PMID: 23468852
- PMCID: PMC3582636
- DOI: 10.1371/journal.pone.0056028
Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury
Abstract
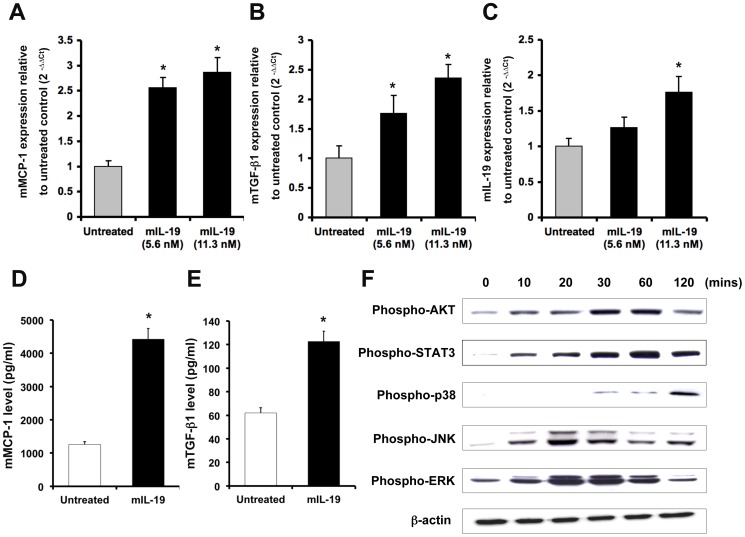
Inflammation and renal tubular injury are major features of acute kidney injury (AKI). Many cytokines and chemokines are released from injured tubular cells and acts as proinflammatory mediators. However, the role of IL-19 in the pathogenesis of AKI is not defined yet. In bilateral renal ischemia/reperfusion injury (IRI)-induced and HgCl2-induced AKI animal models, real-time quantitative (RTQ)-PCR showed that the kidneys, livers, and lungs of AKI mice expressed significantly higher IL-19 and its receptors than did sham control mice. Immunohistochemical staining showed that IL-19 and its receptors were strongly stained in the kidney, liver, and lung tissue of AKI mice. In vitro, IL-19 upregulated MCP-1, TGF-β1, and IL-19, and induced mitochondria-dependent apoptosis in murine renal tubular epithelial M-1 cells. IL-19 upregulated TNF-α and IL-10 in cultured HepG2 cells, and it increased IL-1β and TNF-α expression in cultured A549 cells. In vivo, after renal IRI or a nephrotoxic dose of HgCl2 treatment, IL-20R1-deficient mice (the deficiency blocks IL-19 signaling) showed lower levels of blood urea nitrogen (BUN) in serum and less tubular damage than did wild-type mice. Therefore, we conclude that IL-19 mediates kidney, liver, and lung tissue damage in murine AKI and that blocking IL-19 signaling may provide a potent therapeutic strategy for treating AKI.
Conflict of interest statement
Figures





References
-
- Rana A, Sathyanarayana P, Lieberthal W (2001) Role of apoptosis of renal tubular cells in acute renal failure: therapeutic implications. Apoptosis 6: 83–102. - PubMed
-
- Devarajan P (2006) Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17: 1503–1520. - PubMed
-
- Deng J, Kohda Y, Chiao H, Wang Y, Hu X, et al. (2001) Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int 60: 2118–2128. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
