

Delineating the cytogenomic and epigenomic landscapes of glioma stem cell lines
- PMID: 23468990
- PMCID: PMC3585345
- DOI: 10.1371/journal.pone.0057462
Delineating the cytogenomic and epigenomic landscapes of glioma stem cell lines
Abstract
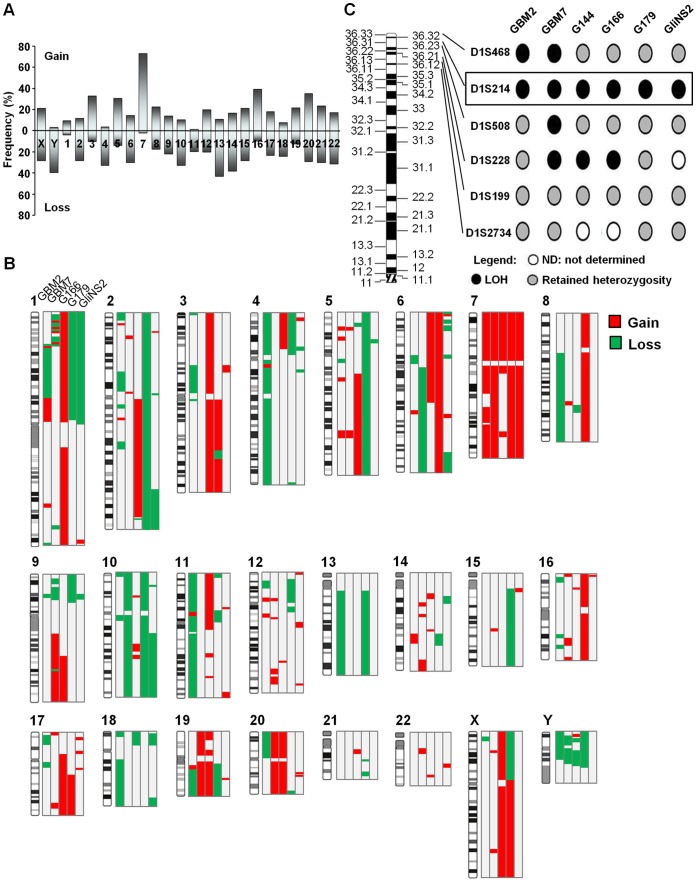
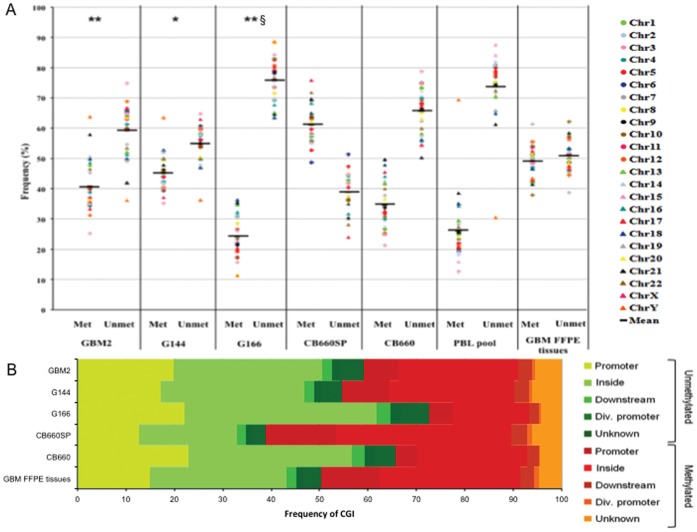
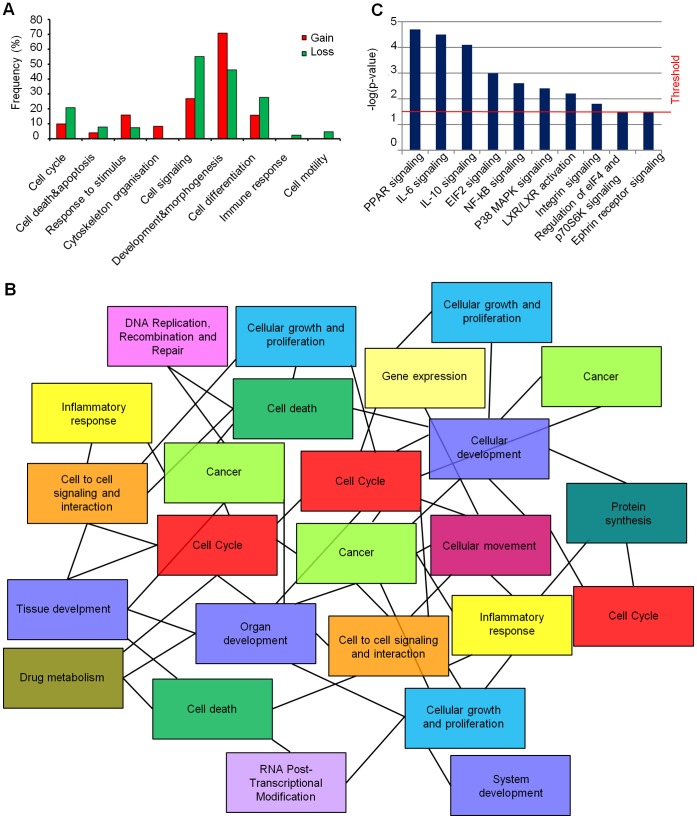
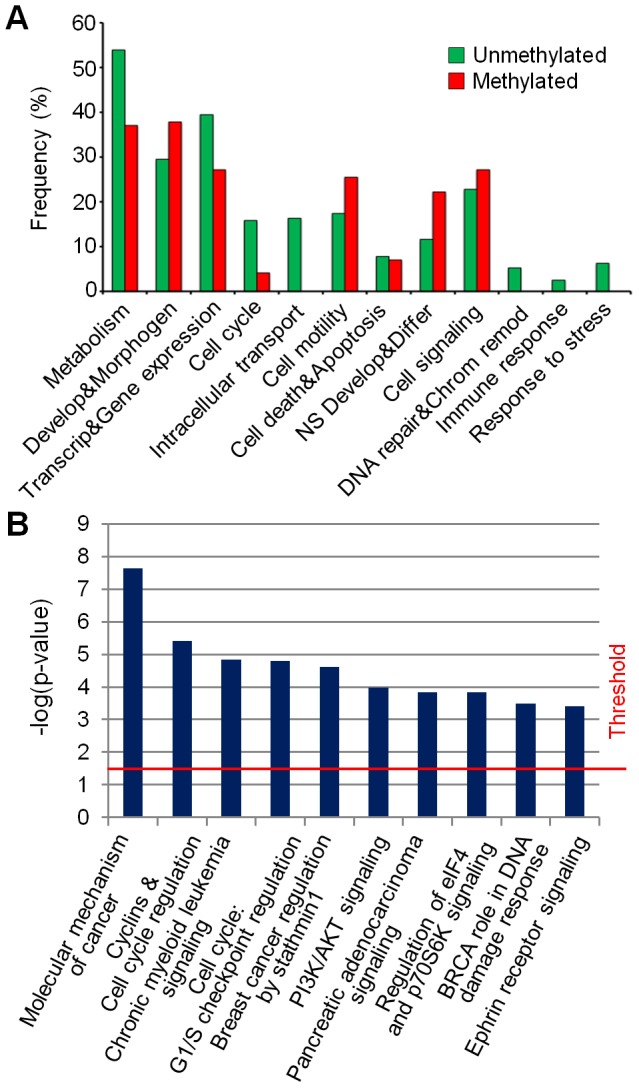
Glioblastoma multiforme (GBM), the most common and malignant type of glioma, is characterized by a poor prognosis and the lack of an effective treatment, which are due to a small sub-population of cells with stem-like properties, termed glioma stem cells (GSCs). The term "multiforme" describes the histological features of this tumor, that is, the cellular and morphological heterogeneity. At the molecular level multiple layers of alterations may reflect this heterogeneity providing together the driving force for tumor initiation and development. In order to decipher the common "signature" of the ancestral GSC population, we examined six already characterized GSC lines evaluating their cytogenomic and epigenomic profiles through a multilevel approach (conventional cytogenetic, FISH, aCGH, MeDIP-Chip and functional bioinformatic analysis). We found several canonical cytogenetic alterations associated with GBM and a common minimal deleted region (MDR) at 1p36.31, including CAMTA1 gene, a putative tumor suppressor gene, specific for the GSC population. Therefore, on one hand our data confirm a role of driver mutations for copy number alterations (CNAs) included in the GBM genomic-signature (gain of chromosome 7- EGFR gene, loss of chromosome 13- RB1 gene, loss of chromosome 10-PTEN gene); on the other, it is not obvious that the new identified CNAs are passenger mutations, as they may be necessary for tumor progression specific for the individual patient. Through our approach, we were able to demonstrate that not only individual genes into a pathway can be perturbed through multiple mechanisms and at different levels, but also that different combinations of perturbed genes can incapacitate functional modules within a cellular networks. Therefore, beyond the differences that can create apparent heterogeneity of alterations among GSC lines, there's a sort of selective force acting on them in order to converge towards the impairment of cell development and differentiation processes. This new overview could have a huge importance in therapy.
Conflict of interest statement
Figures

References
-
- Hegi ME, Janzer RC, Lambiv WL, Gorlia T, Kouwenhoven MC, et al. (2012) Presence of an oligodendroglioma-like component in newly diagnosed glioblastoma identifies a pathogenetically heterogeneous subgroup and lacks prognostic value: central pathology review of the EORTC_26981/NCIC_CE.3 trial. Acta Neuropathol 123: 841–852. - PubMed
-
- Gorlia T, Stupp R, Brandes AA, Rampling RR, Fumoleau P, et al. (2012) New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer 48: 1176–1184. - PubMed
-
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996. - PubMed
-
- Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359: 492–507. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
