

Newborn screening 50 years later: access issues faced by adults with PKU
- PMID: 23470838
- PMCID: PMC3938172
- DOI: 10.1038/gim.2013.10
Newborn screening 50 years later: access issues faced by adults with PKU
Abstract
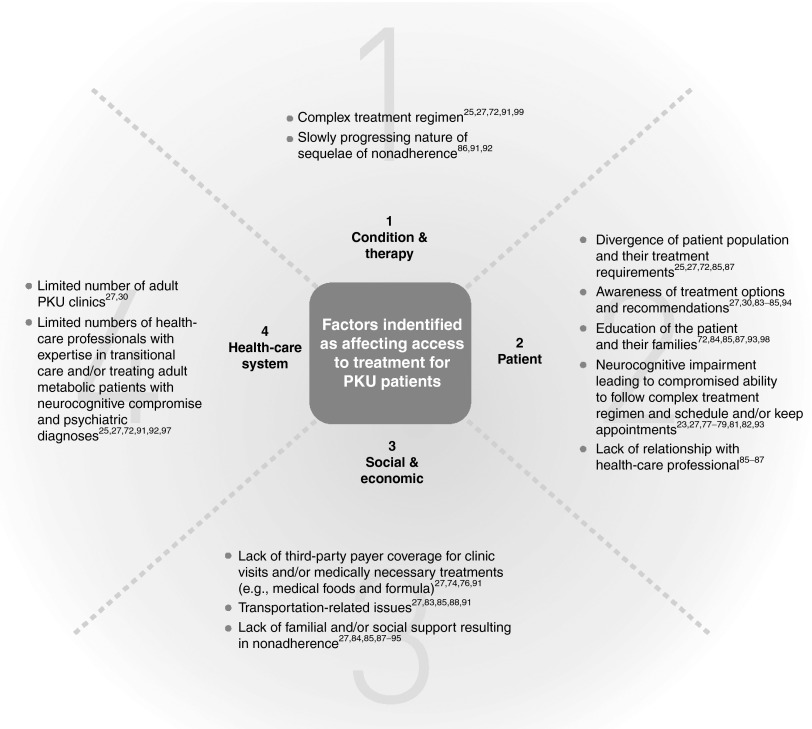
Fifty years after the implementation of universal newborn screening programs for phenylketonuria, the first disease identified through newborn screening and considered a success story of newborn screening, a cohort of adults with phenylketonuria treated from birth provides valuable information about effects of long-term treatment for inborn errors of metabolism in general, and phenylketonuria specifically. For phenylketonuria, newborn screening allows early implementation of the phenylalanine-restricted diet, eliminating the severe neurocognitive and neuromotor impairment associated with untreated phenylketonuria. However, executive function impairments and psychiatric problems are frequently reported even for those treated early and continuously with the phenylalanine-restricted diet alone. Moreover, a large percentage of adults with phenylketonuria are reported as lost to follow-up by metabolic clinics. While a group of experts identified by the National Institutes of Health convenes to update treatment guidelines for phenylketonuria, we explore individual patient, social, and economic factors preventing >70% of adult phenylketonuria patients in the United States from accessing treatment. As more conditions are identified through newborn screening, factors affecting access to treatment grow in importance, and we must continue to be vigilant in assessing and addressing factors that affect patient treatment outcomes and not just celebrate amelioration of the most severe manifestations of disease.
Figures
References
-
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–343. - PubMed
-
- Folling A. The excretion of phenylpyruvic acid in the urine, an anomaly of metabolism in connection with imbecility. Zeitschrift fur PhysiologischeChemie. 1934;227:169–176.
-
- Scriver CR, Levy H, Donlon J.Hyperphenylalaninemia: phenylalanine hydroxylase deficiency Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A.eds). The online metabolic and molecular bases of inherited disease 2008 . http://www.ommbid.com .
-
- Koch R, Dobson JC. Hospital screening programs aid identification of PKU. Hosp Top. 1968;46:111–113. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
