

Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy
- PMID: 23473319
- PMCID: PMC3594792
- DOI: 10.1016/j.neuron.2013.01.012
Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy
Abstract
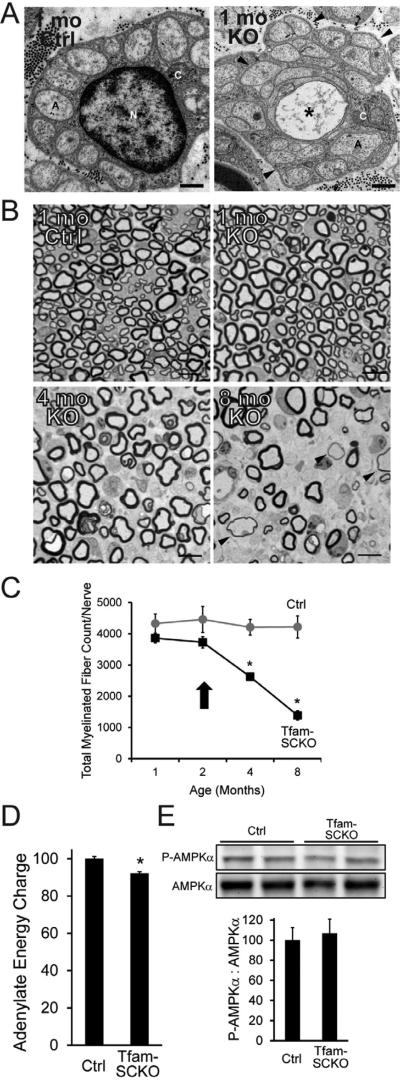
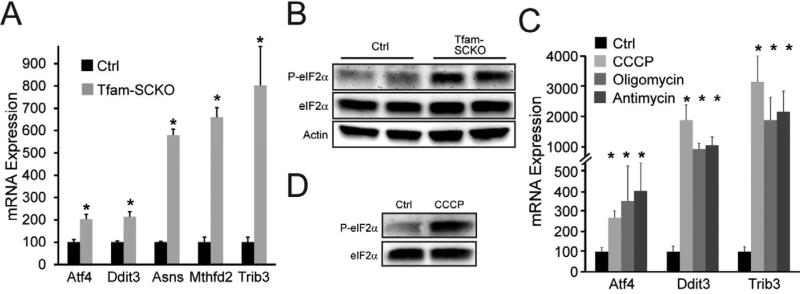
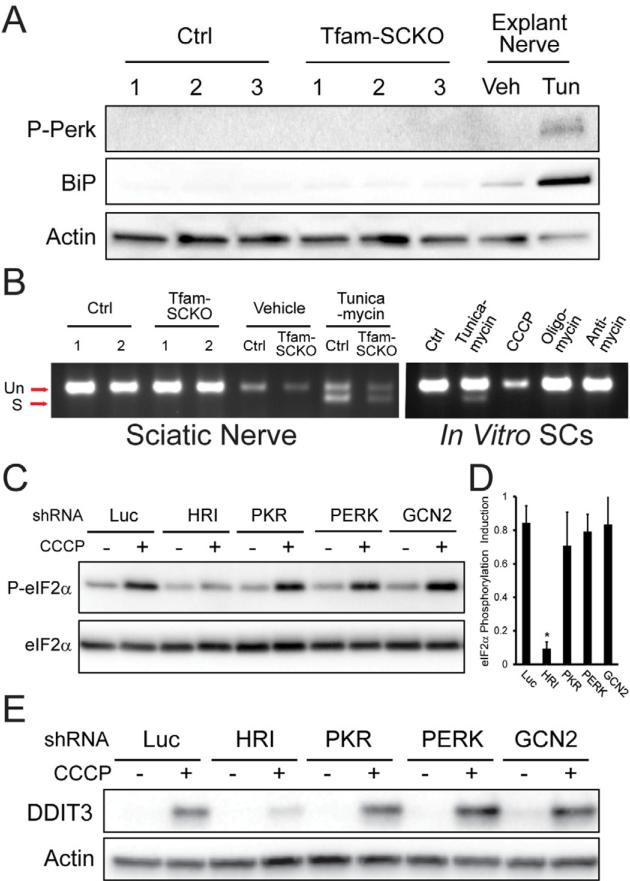
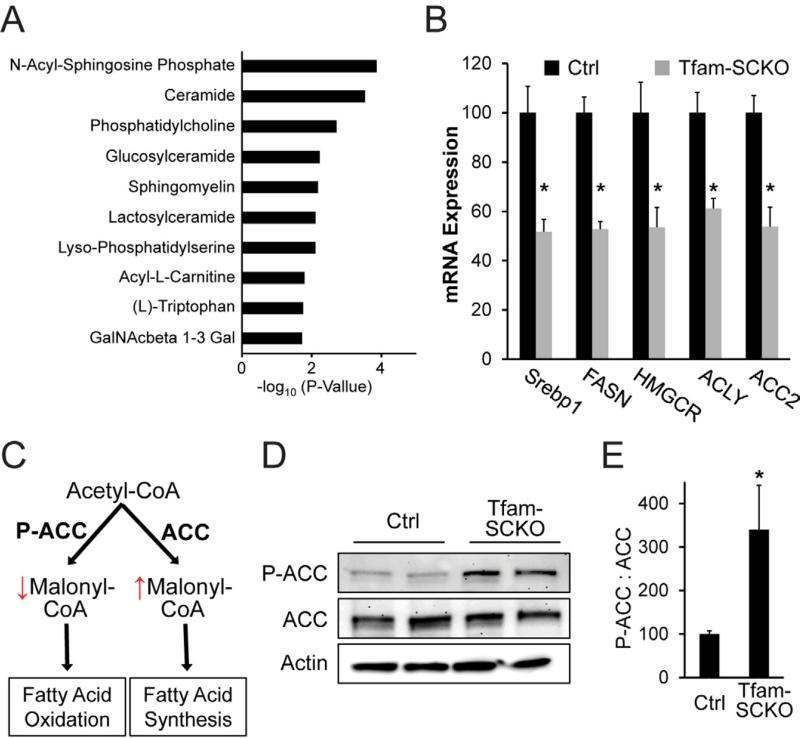
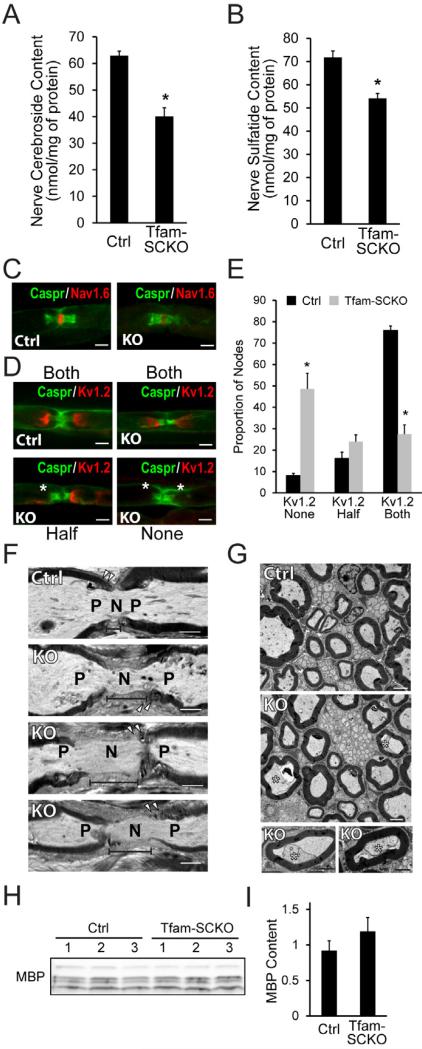
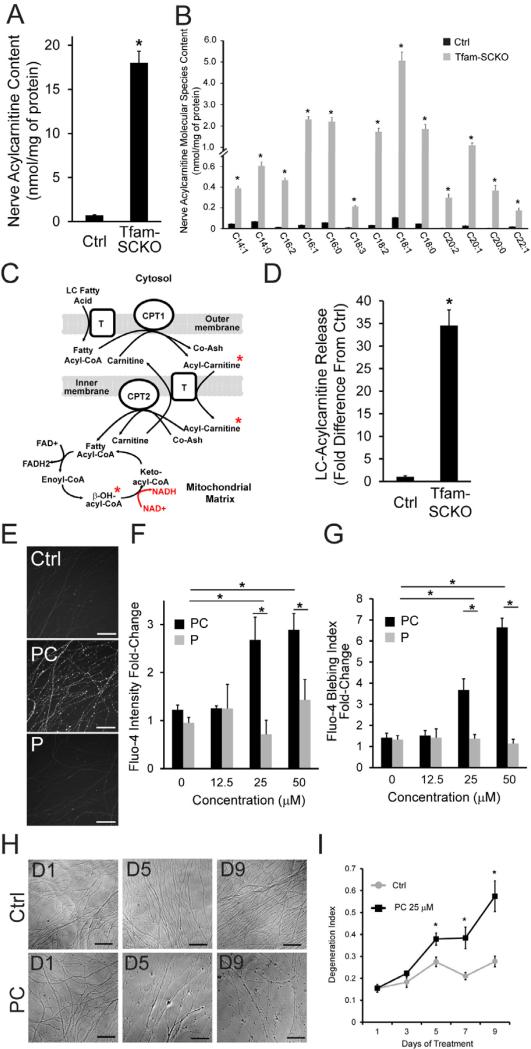
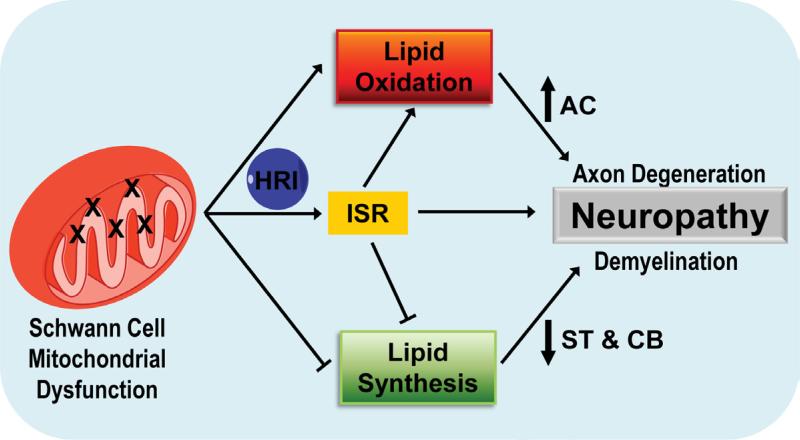
Mitochondrial dysfunction is a common cause of peripheral neuropathy. Much effort has been devoted to examining the role played by neuronal/axonal mitochondria, but how mitochondrial deficits in peripheral nerve glia (Schwann cells [SCs]) contribute to peripheral nerve diseases remains unclear. Here, we investigate a mouse model of peripheral neuropathy secondary to SC mitochondrial dysfunction (Tfam-SCKOs). We show that disruption of SC mitochondria activates a maladaptive integrated stress response (ISR) through the actions of heme-regulated inhibitor (HRI) kinase, and causes a shift in lipid metabolism away from fatty acid synthesis toward oxidation. These alterations in SC lipid metabolism result in depletion of important myelin lipid components as well as in accumulation of acylcarnitines (ACs), an intermediate of fatty acid β-oxidation. Importantly, we show that ACs are released from SCs and induce axonal degeneration. A maladaptive ISR as well as altered SC lipid metabolism are thus underlying pathological mechanisms in mitochondria-related peripheral neuropathies.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
When Schwann cells conspire with mitochondria, neighboring axons are under attack by glia-derived neurotoxic lipids.Neuron. 2013 Mar 6;77(5):801-3. doi: 10.1016/j.neuron.2013.02.016. Neuron. 2013. PMID: 23473310
References
-
- Atkinson DE. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry. 1968;7:4030–4034. - PubMed
-
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. - PubMed
-
- Barber MC, Price NT, Travers MT. Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim Biophys Acta. 2005;1733:1–28. - PubMed
-
- Barres B. a. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- AG13730/AG/NIA NIH HHS/United States
- P30 NS057105/NS/NINDS NIH HHS/United States
- RF1 AG013730/AG/NIA NIH HHS/United States
- R01 AG013730/AG/NIA NIH HHS/United States
- P30 CA091842/CA/NCI NIH HHS/United States
- 2P01 HL057278/HL/NHLBI NIH HHS/United States
- R56 AG013730/AG/NIA NIH HHS/United States
- P30 DK020579/DK/NIDDK NIH HHS/United States
- P01 HL057278/HL/NHLBI NIH HHS/United States
- UL1RR024992/RR/NCRR NIH HHS/United States
- UL1 RR024992/RR/NCRR NIH HHS/United States
- P30 CA91842/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
