

A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies
- PMID: 23478172
- PMCID: PMC4103162
- DOI: 10.1016/j.nmd.2013.02.009
A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies
Abstract
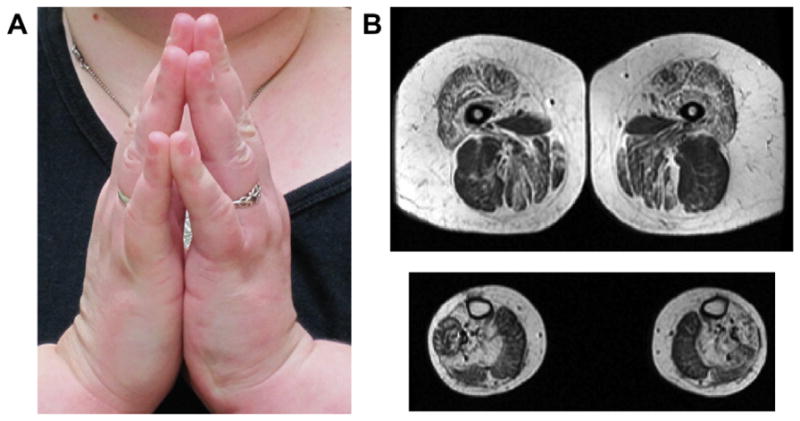
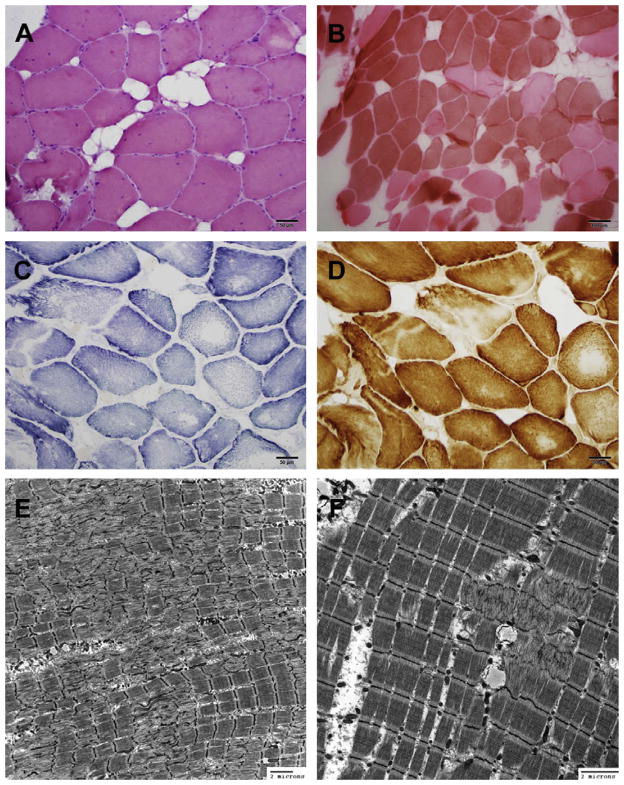
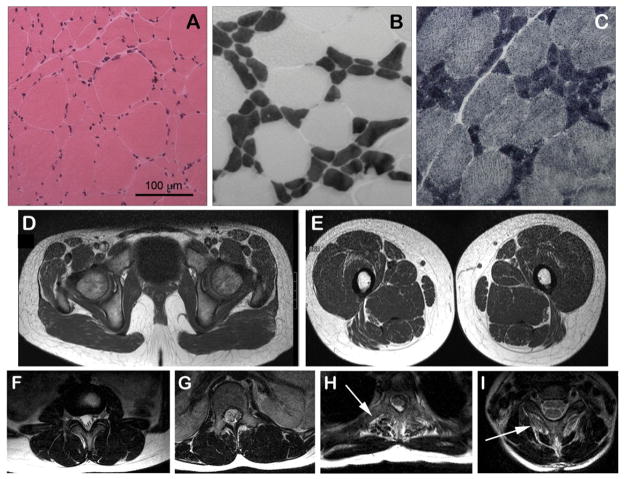
MYH7 mutations are an established cause of Laing distal myopathy, myosin storage myopathy, and cardiomyopathy, as well as additional myopathy subtypes. We report a novel MYH7 mutation (p.Leu1597Arg) that arose de novo in two unrelated probands. Proband 1 has a myopathy characterized by distal weakness and prominent contractures and histopathology typical of multi-minicore disease. Proband 2 has an axial myopathy and histopathology consistent with congenital fiber type disproportion. These cases highlight the broad spectrum of clinical and histological patterns associated with MYH7 mutations, and provide further evidence that MYH7 is likely responsible for a greater proportion of congenital myopathies than currently appreciated.
Copyright © 2013 Elsevier B.V. All rights reserved.
Figures
References
-
- Engel A, Franzini-Armstrong C. Myology: basic and clinical. 3. New York: McGraw-Hill, Medical Pub. Division; 2004.
-
- Capek PC. Gene symbol: MYH7. Disease: cardiomyopathy, hypertrophic. Hum Genet. 2005;118(3–4):537. - PubMed
-
- Dye DE, Azzarelli B, Goebel HH, Laing NG. Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul Disord. 2006;16(6):357–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
