

The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes
- PMID: 23478400
- PMCID: PMC3638132
- DOI: 10.1101/gr.148718.112
The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes
Abstract
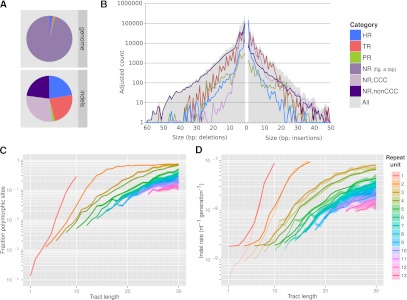
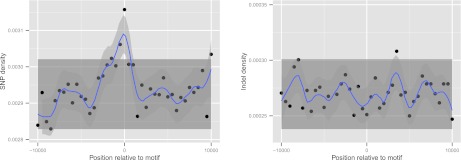
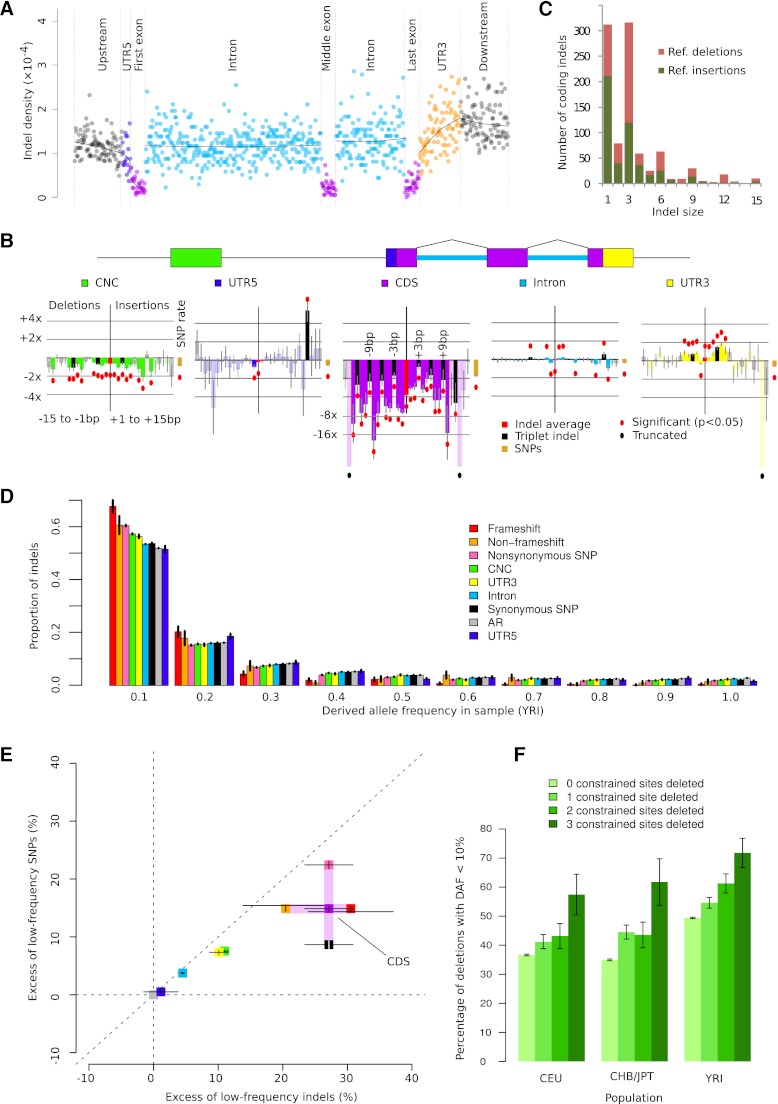
Short insertions and deletions (indels) are the second most abundant form of human genetic variation, but our understanding of their origins and functional effects lags behind that of other types of variants. Using population-scale sequencing, we have identified a high-quality set of 1.6 million indels from 179 individuals representing three diverse human populations. We show that rates of indel mutagenesis are highly heterogeneous, with 43%-48% of indels occurring in 4.03% of the genome, whereas in the remaining 96% their prevalence is 16 times lower than SNPs. Polymerase slippage can explain upwards of three-fourths of all indels, with the remainder being mostly simple deletions in complex sequence. However, insertions do occur and are significantly associated with pseudo-palindromic sequence features compatible with the fork stalling and template switching (FoSTeS) mechanism more commonly associated with large structural variations. We introduce a quantitative model of polymerase slippage, which enables us to identify indel-hypermutagenic protein-coding genes, some of which are associated with recurrent mutations leading to disease. Accounting for mutational rate heterogeneity due to sequence context, we find that indels across functional sequence are generally subject to stronger purifying selection than SNPs. We find that indel length modulates selection strength, and that indels affecting multiple functionally constrained nucleotides undergo stronger purifying selection. We further find that indels are enriched in associations with gene expression and find evidence for a contribution of nonsense-mediated decay. Finally, we show that indels can be integrated in existing genome-wide association studies (GWAS); although we do not find direct evidence that potentially causal protein-coding indels are enriched with associations to known disease-associated SNPs, our findings suggest that the causal variant underlying some of these associations may be indels.
Figures

References
-
- Bhangale TR, Rieder MJ, Livingston RJ, Nickerson DA 2005. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Hum Mol Genet 14: 59–69 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources