

Synaptic plasticity by antidromic firing during hippocampal network oscillations
- PMID: 23479613
- PMCID: PMC3612622
- DOI: 10.1073/pnas.1210735110
Synaptic plasticity by antidromic firing during hippocampal network oscillations
Abstract
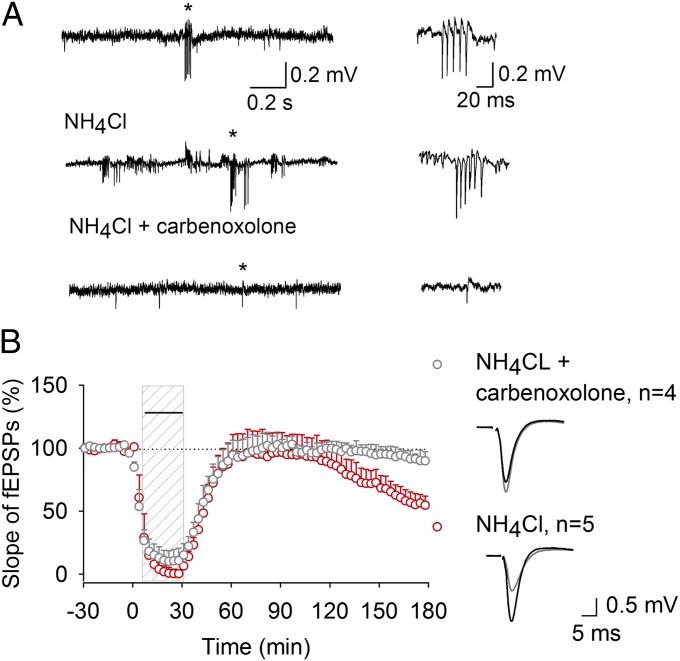
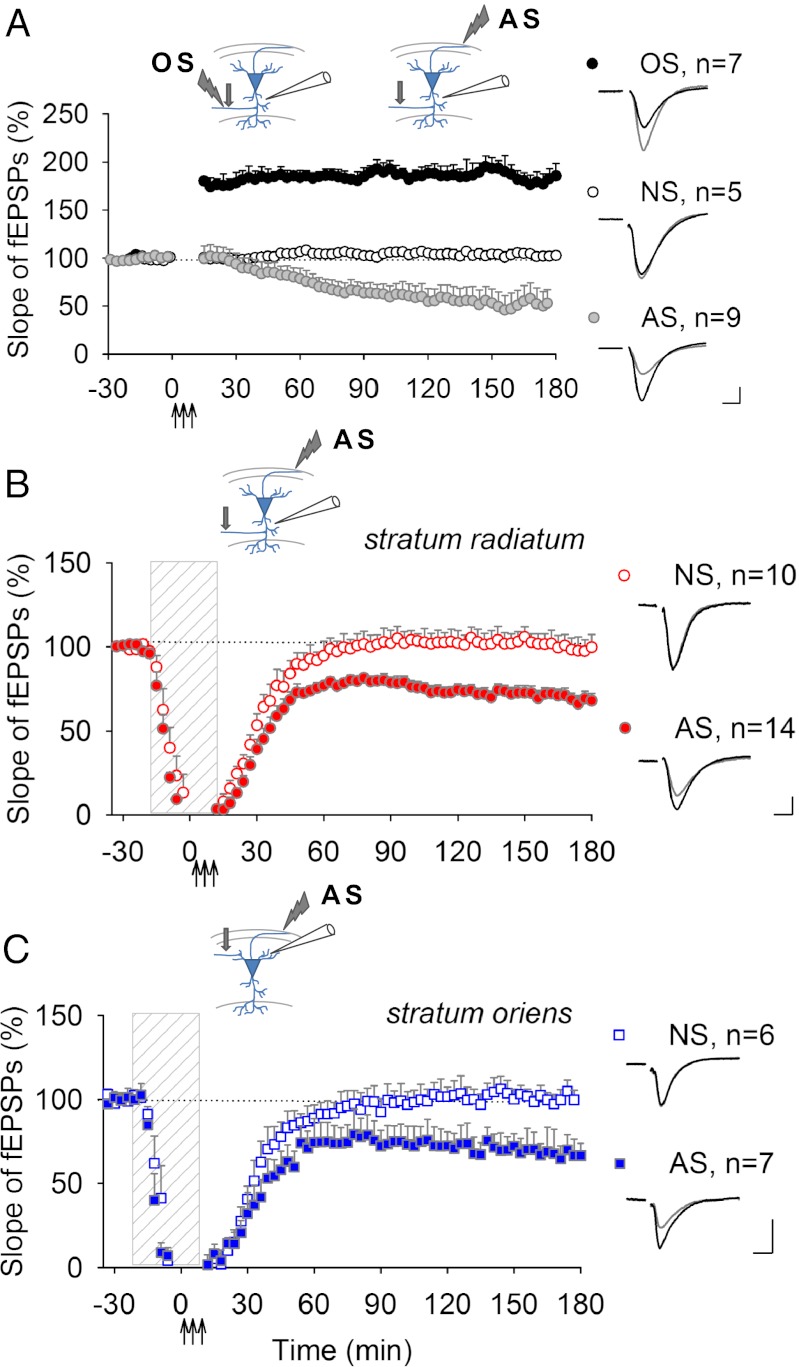
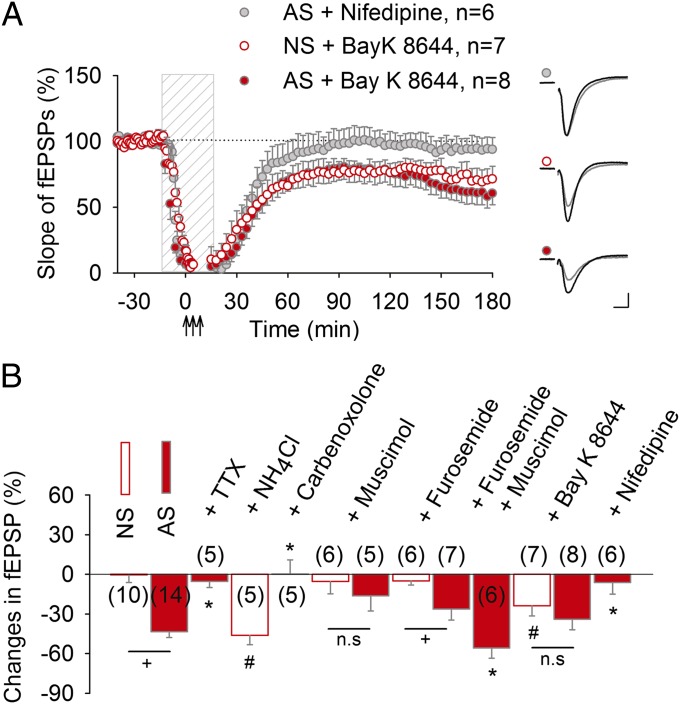
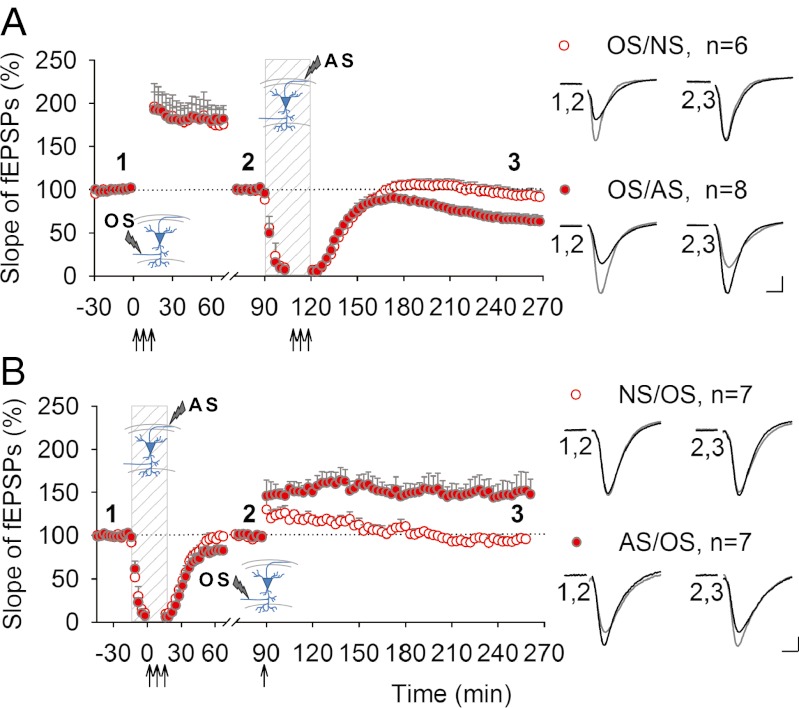
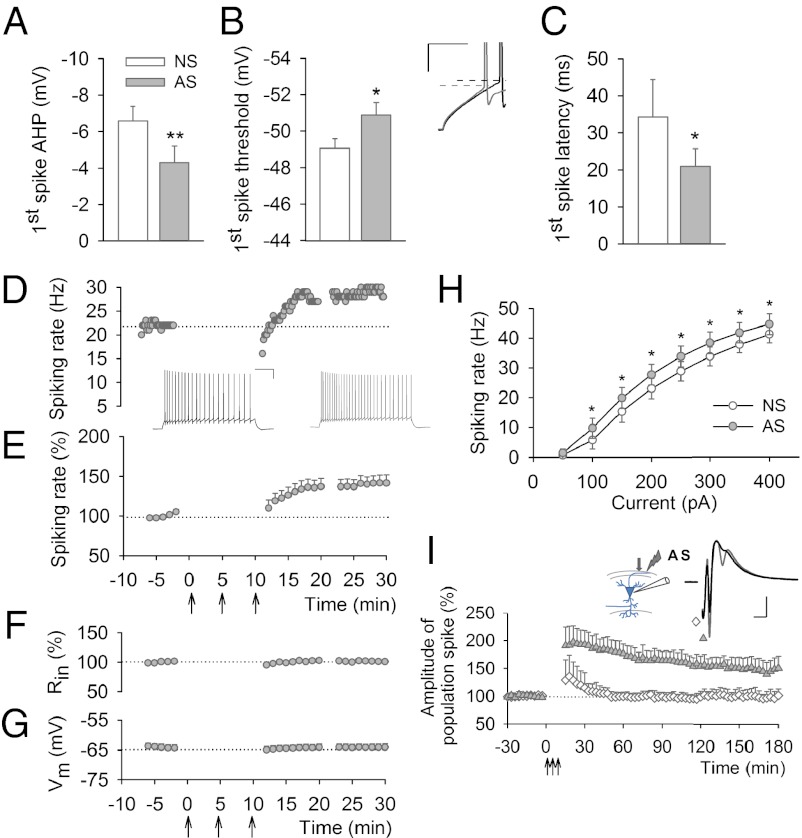
Learning and other cognitive tasks require integrating new experiences into context. In contrast to sensory-evoked synaptic plasticity, comparatively little is known of how synaptic plasticity may be regulated by intrinsic activity in the brain, much of which can involve nonclassical modes of neuronal firing and integration. Coherent high-frequency oscillations of electrical activity in CA1 hippocampal neurons [sharp-wave ripple complexes (SPW-Rs)] functionally couple neurons into transient ensembles. These oscillations occur during slow-wave sleep or at rest. Neurons that participate in SPW-Rs are distinguished from adjacent nonparticipating neurons by firing action potentials that are initiated ectopically in the distal region of axons and propagate antidromically to the cell body. This activity is facilitated by GABA(A)-mediated depolarization of axons and electrotonic coupling. The possible effects of antidromic firing on synaptic strength are unknown. We find that facilitation of spontaneous SPW-Rs in hippocampal slices by increasing gap-junction coupling or by GABA(A)-mediated axon depolarization resulted in a reduction of synaptic strength, and electrical stimulation of axons evoked a widespread, long-lasting synaptic depression. Unlike other forms of synaptic plasticity, this synaptic depression is not dependent upon synaptic input or glutamate receptor activation, but rather requires L-type calcium channel activation and functional gap junctions. Synaptic stimulation delivered after antidromic firing, which was otherwise too weak to induce synaptic potentiation, triggered a long-lasting increase in synaptic strength. Rescaling synaptic weights in subsets of neurons firing antidromically during SPW-Rs might contribute to memory consolidation by sharpening specificity of subsequent synaptic input and promoting incorporation of novel information.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Buzsáki G. Two-stage model of memory trace formation: A role for “noisy” brain states. Neuroscience. 1989;31(3):551–570. - PubMed
-
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265(5172):676–679. - PubMed
-
- Lewis PA, Durrant SJ. Overlapping memory replay during sleep builds cognitive schemata. Trends Cogn Sci. 2011;15(8):343–351. - PubMed
-
- Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nat Rev Neurosci. 2000;1(3):216–219. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
