

Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability
- PMID: 23486205
- PMCID: PMC3680799
- DOI: 10.1152/jn.00500.2012
Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability
Abstract
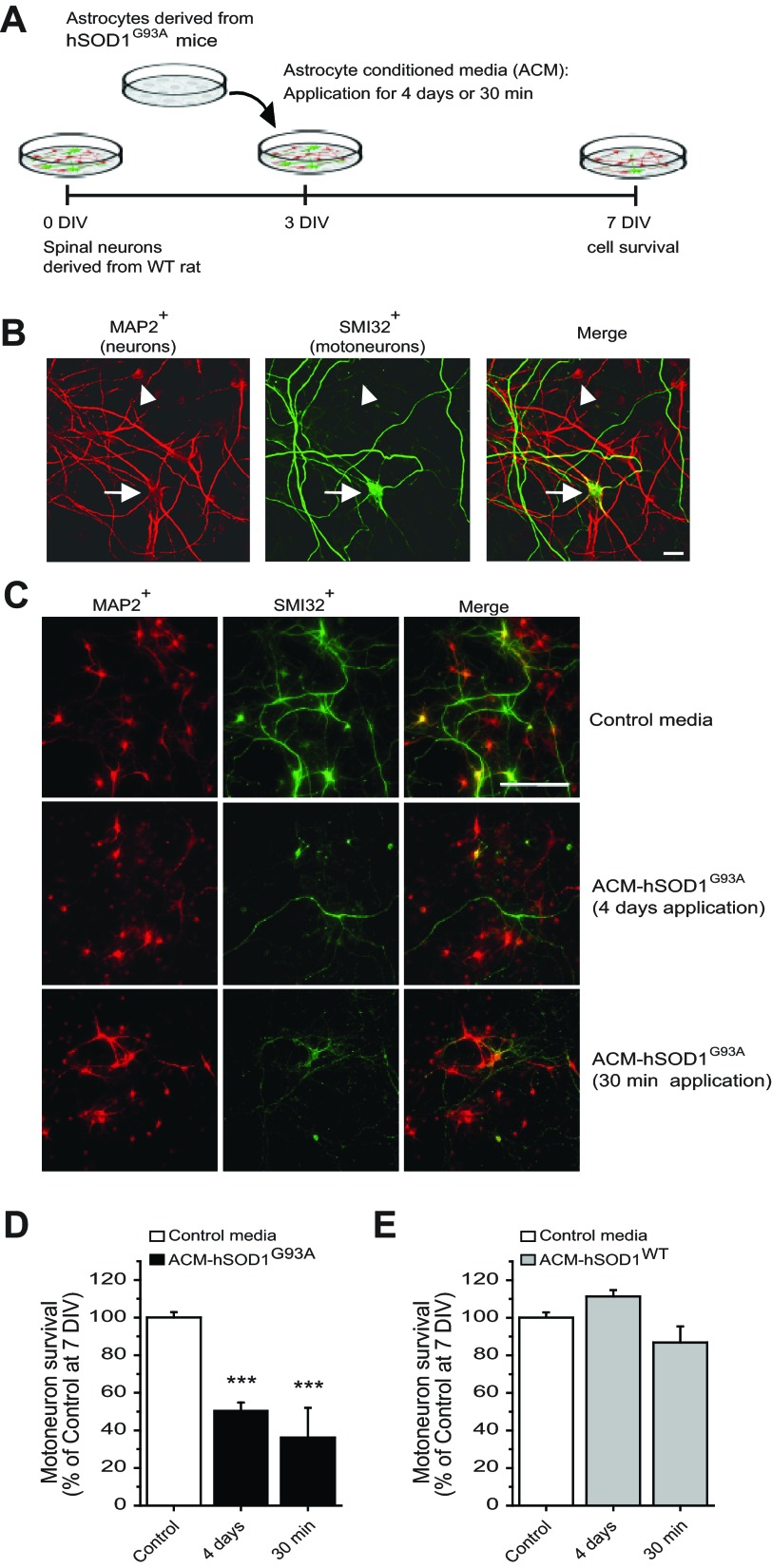
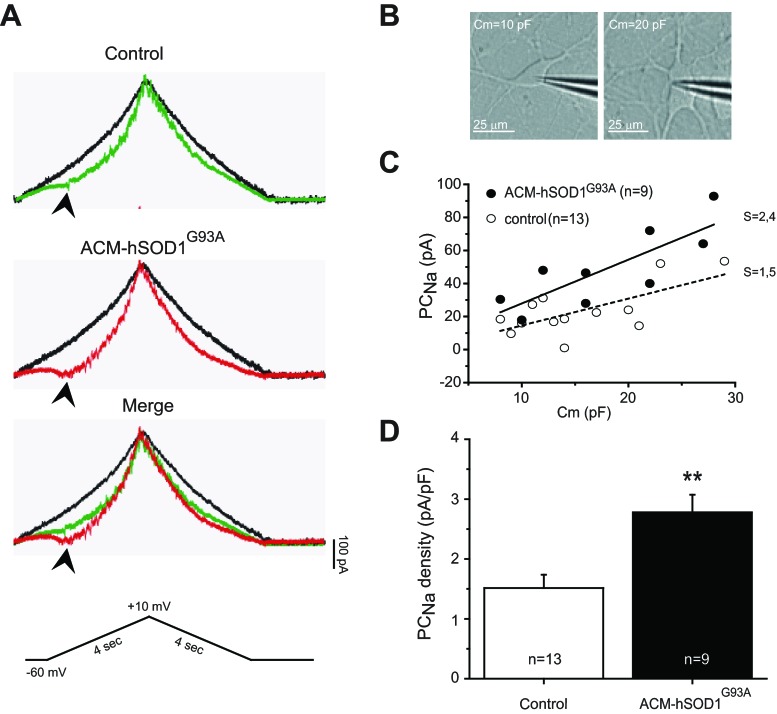
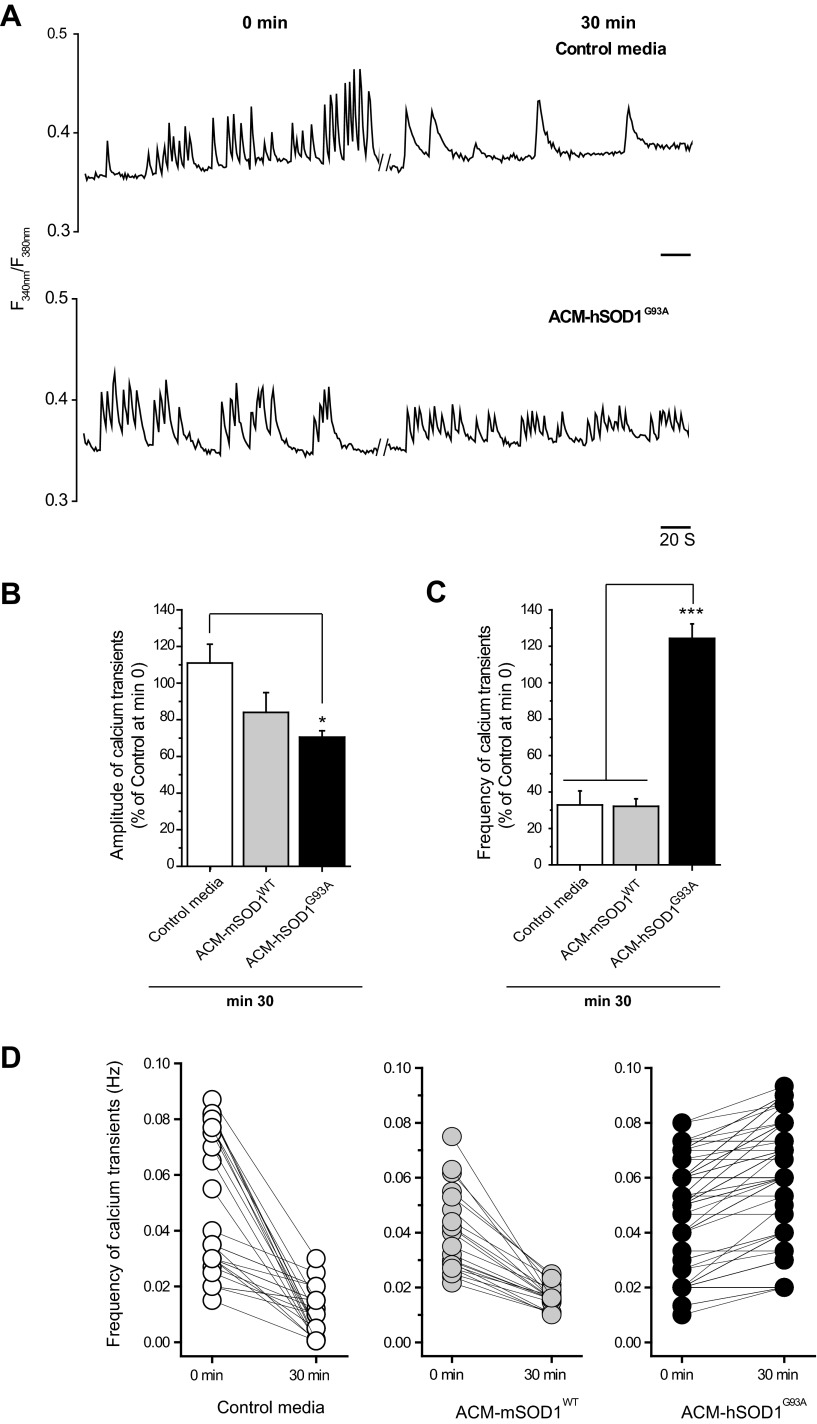
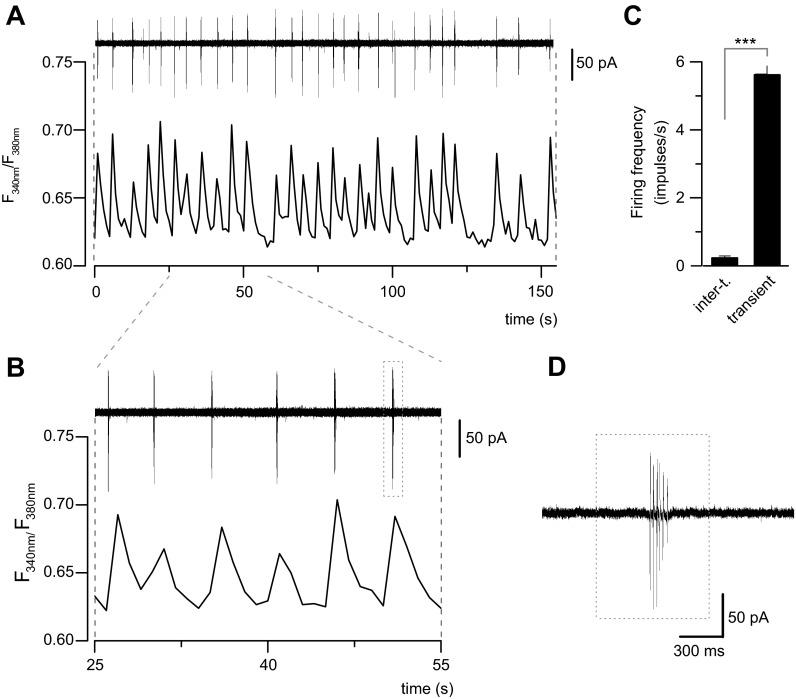
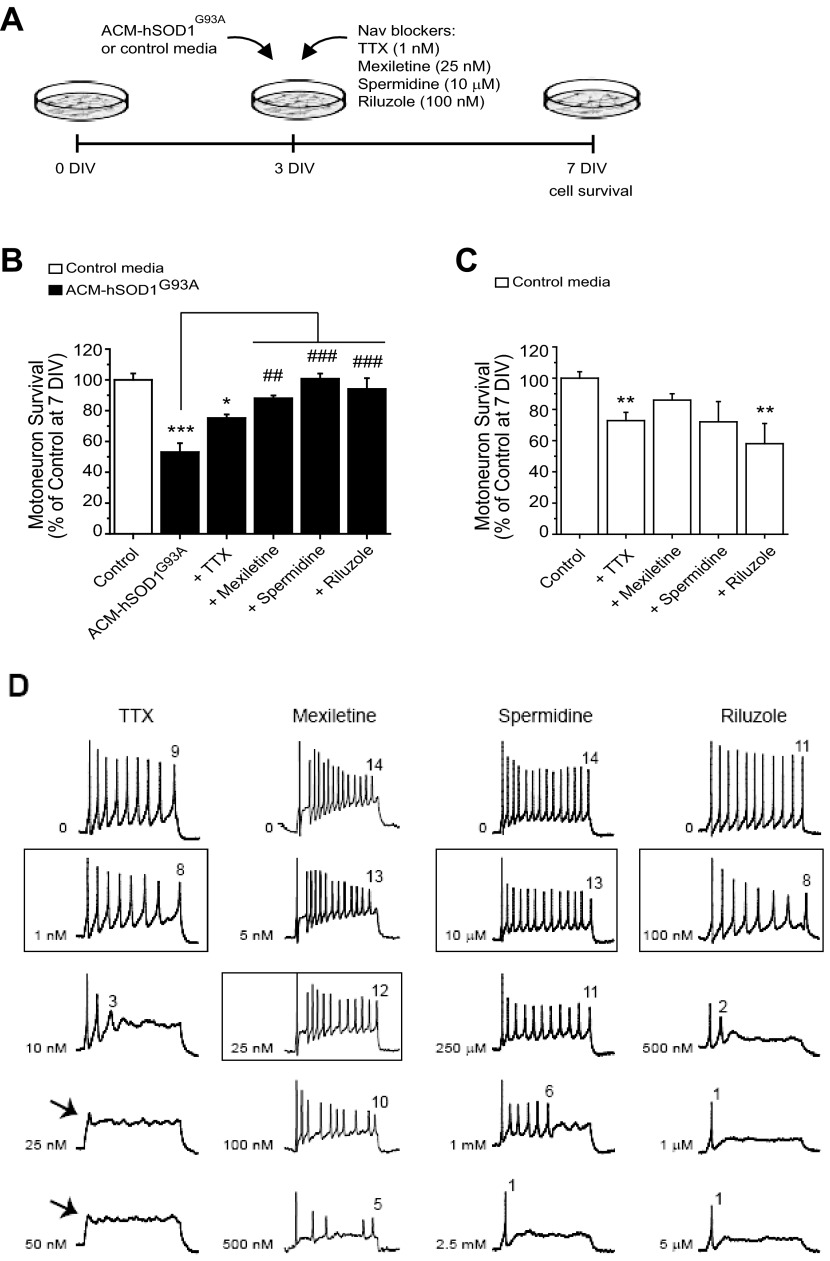
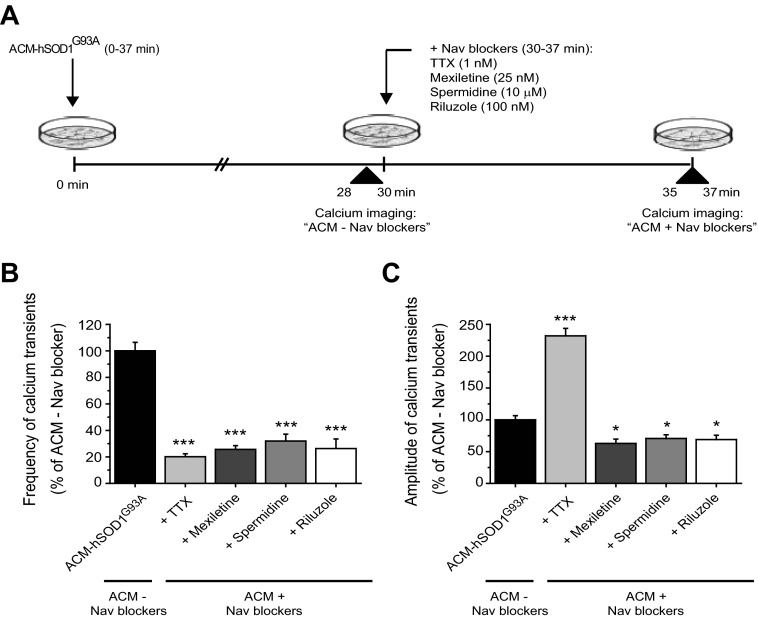
Amyotrophic lateral sclerosis (ALS) is a devastating paralytic disorder caused by dysfunction and degeneration of motoneurons starting in adulthood. Recent studies using cell or animal models document that astrocytes expressing disease-causing mutations of human superoxide dismutase 1 (hSOD1) contribute to the pathogenesis of ALS by releasing a neurotoxic factor(s). Neither the mechanism by which this neurotoxic factor induces motoneuron death nor its cellular site of action has been elucidated. Here we show that acute exposure of primary wild-type spinal cord cultures to conditioned medium derived from astrocytes expressing mutant SOD1 (ACM-hSOD1(G93A)) increases persistent sodium inward currents (PC(Na)), repetitive firing, and intracellular calcium transients, leading to specific motoneuron death days later. In contrast to TTX, which paradoxically increased twofold the amplitude of calcium transients and killed motoneurons, reduction of hyperexcitability by other specific (mexiletine) and nonspecific (spermidine and riluzole) blockers of voltage-sensitive sodium (Na(v)) channels restored basal calcium transients and prevented motoneuron death induced by ACM-hSOD1(G93A). These findings suggest that riluzole, the only FDA-approved drug with known benefits for ALS patients, acts by inhibiting hyperexcitability. Together, our data document that a critical element mediating the non-cell-autonomous toxicity of ACM-hSOD1(G93A) on motoneurons is increased excitability, an observation with direct implications for therapy of ALS.
Keywords: amyotrophic lateral sclerosis; hyperexcitability; motoneuron degeneration; sodium channel.
Figures

Similar articles
-
Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress.Front Cell Neurosci. 2014 Feb 7;8:24. doi: 10.3389/fncel.2014.00024. eCollection 2014. Front Cell Neurosci. 2014. PMID: 24570655 Free PMC article.
-
Novel Dual Mechanism GRT-X Agonist Acting on Kv7 Potassium Channel/Translocator Protein Receptor Prevents Motoneuron Degeneration Following Exposure to Mouse and Human Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Astrocyte-Conditioned Media.ACS Chem Neurosci. 2025 Aug 6;16(15):2887-2900. doi: 10.1021/acschemneuro.5c00197. Epub 2025 Jul 17. ACS Chem Neurosci. 2025. PMID: 40671688 Free PMC article.
-
Chronic infusion of SOD1G93A astrocyte-secreted factors induces spinal motoneuron degeneration and neuromuscular dysfunction in healthy rats.J Cell Physiol. 2017 Oct;232(10):2610-2615. doi: 10.1002/jcp.25827. Epub 2017 Mar 27. J Cell Physiol. 2017. PMID: 28128448
-
Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis.Mol Neurobiol. 2012 Feb;45(1):30-42. doi: 10.1007/s12035-011-8217-x. Epub 2011 Nov 10. Mol Neurobiol. 2012. PMID: 22072396 Free PMC article. Review.
-
[Proximal axonal injuries as experimental models for adult motoneuron degeneration].Brain Nerve. 2007 Oct;59(10):1179-86. Brain Nerve. 2007. PMID: 17969359 Review. Japanese.
Cited by
-
Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress.Front Cell Neurosci. 2014 Feb 7;8:24. doi: 10.3389/fncel.2014.00024. eCollection 2014. Front Cell Neurosci. 2014. PMID: 24570655 Free PMC article.
-
Axonal Excitability in Amyotrophic Lateral Sclerosis : Axonal Excitability in ALS.Neurotherapeutics. 2017 Jan;14(1):78-90. doi: 10.1007/s13311-016-0492-9. Neurotherapeutics. 2017. PMID: 27878516 Free PMC article. Review.
-
Emerging Mechanisms Underpinning Neurophysiological Impairments in C9ORF72 Repeat Expansion-Mediated Amyotrophic Lateral Sclerosis/Frontotemporal Dementia.Front Cell Neurosci. 2021 Dec 15;15:784833. doi: 10.3389/fncel.2021.784833. eCollection 2021. Front Cell Neurosci. 2021. PMID: 34975412 Free PMC article. Review.
-
Mature iPSC-derived astrocytes of an ALS/FTD patient carrying the TDP43A90V mutation display a mild reactive state and release polyP toxic to motoneurons.Front Cell Dev Biol. 2023 Jul 28;11:1226604. doi: 10.3389/fcell.2023.1226604. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37645251 Free PMC article.
-
Regulation of cortical hyperexcitability in amyotrophic lateral sclerosis: focusing on glial mechanisms.Mol Neurodegener. 2023 Oct 19;18(1):75. doi: 10.1186/s13024-023-00665-w. Mol Neurodegener. 2023. PMID: 37858176 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous