

Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis
- PMID: 23486940
- PMCID: PMC3711648
- DOI: 10.1523/JNEUROSCI.1119-12.2013
Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis
Abstract
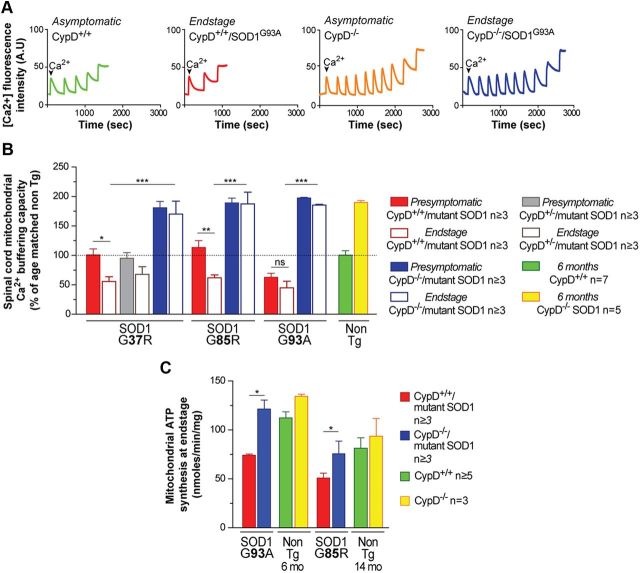
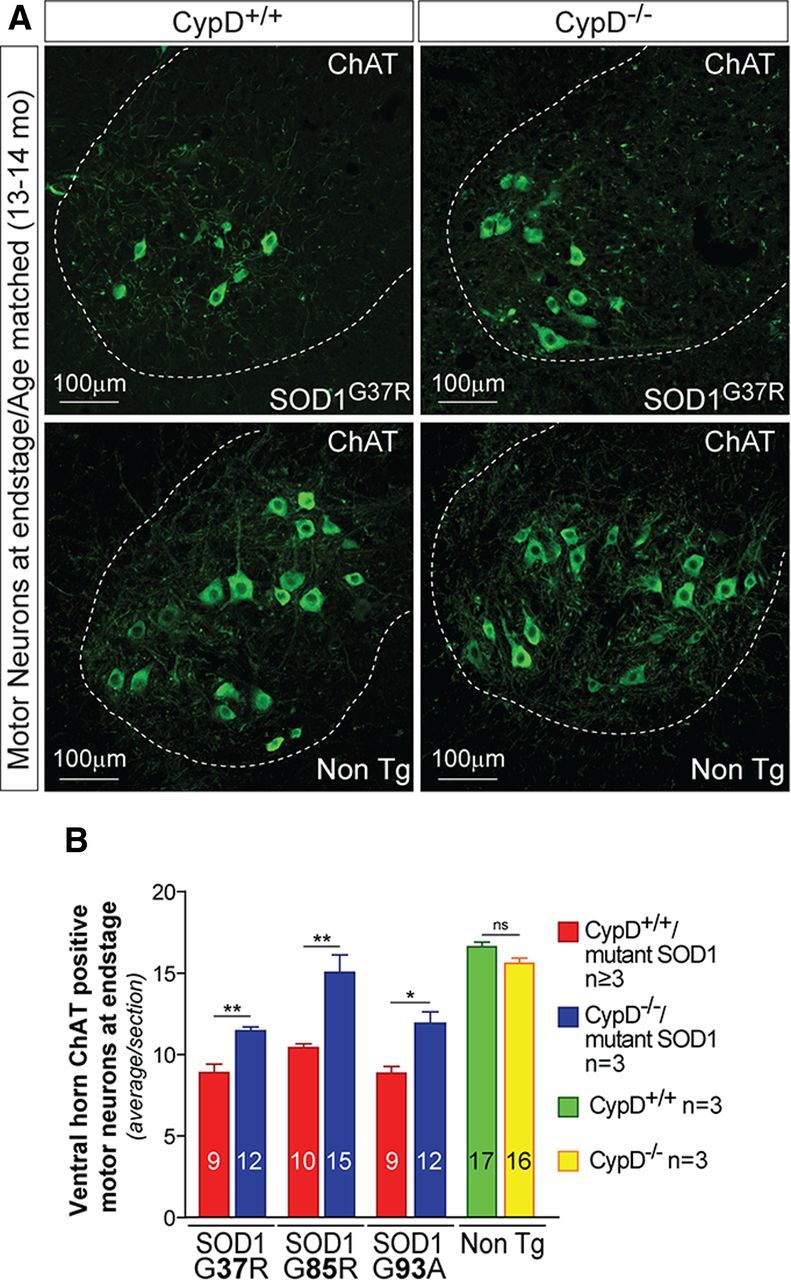
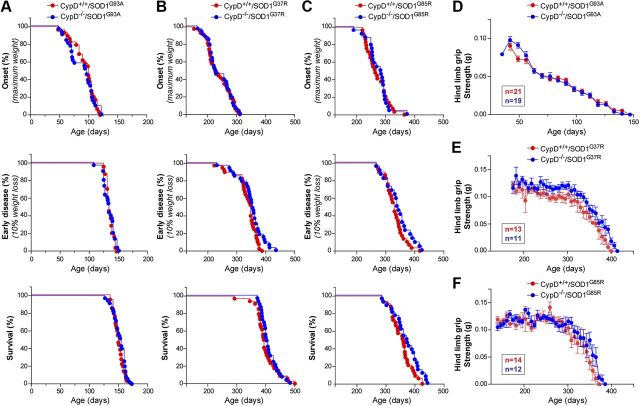
Mitochondria have been proposed as targets for toxicity in amyotrophic lateral sclerosis (ALS), a progressive, fatal adult-onset neurodegenerative disorder characterized by the selective loss of motor neurons. A decrease in the capacity of spinal cord mitochondria to buffer calcium (Ca(2+)) has been observed in mice expressing ALS-linked mutants of SOD1 that develop motor neuron disease with many of the key pathological hallmarks seen in ALS patients. In mice expressing three different ALS-causing SOD1 mutants, we now test the contribution of the loss of mitochondrial Ca(2+)-buffering capacity to disease mechanism(s) by eliminating ubiquitous expression of cyclophilin D, a critical regulator of Ca(2+)-mediated opening of the mitochondrial permeability transition pore that determines mitochondrial Ca(2+) content. A chronic increase in mitochondrial buffering of Ca(2+) in the absence of cyclophilin D was maintained throughout disease course and was associated with improved mitochondrial ATP synthesis, reduced mitochondrial swelling, and retention of normal morphology. This was accompanied by an attenuation of glial activation, reduction in levels of misfolded SOD1 aggregates in the spinal cord, and a significant suppression of motor neuron death throughout disease. Despite this, muscle denervation, motor axon degeneration, and disease progression and survival were unaffected, thereby eliminating mutant SOD1-mediated loss of mitochondrial Ca(2+) buffering capacity, altered mitochondrial morphology, motor neuron death, and misfolded SOD1 aggregates, as primary contributors to disease mechanism for fatal paralysis in these models of familial ALS.
Figures






References
-
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. - PubMed
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. - PubMed
-
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous