

Make the most of your samples: Bayes factor estimators for high-dimensional models of sequence evolution
- PMID: 23497171
- PMCID: PMC3651733
- DOI: 10.1186/1471-2105-14-85
Make the most of your samples: Bayes factor estimators for high-dimensional models of sequence evolution
Abstract
Background: Accurate model comparison requires extensive computation times, especially for parameter-rich models of sequence evolution. In the Bayesian framework, model selection is typically performed through the evaluation of a Bayes factor, the ratio of two marginal likelihoods (one for each model). Recently introduced techniques to estimate (log) marginal likelihoods, such as path sampling and stepping-stone sampling, offer increased accuracy over the traditional harmonic mean estimator at an increased computational cost. Most often, each model's marginal likelihood will be estimated individually, which leads the resulting Bayes factor to suffer from errors associated with each of these independent estimation processes.
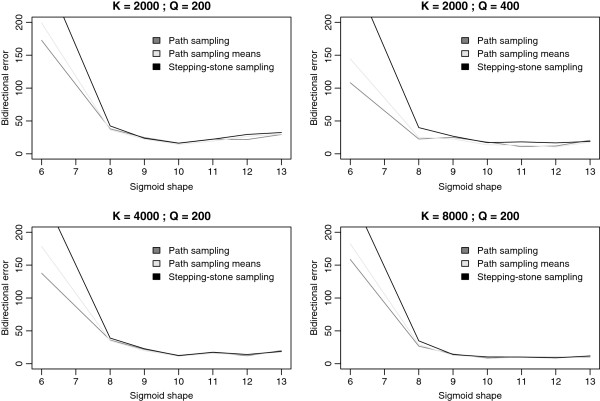
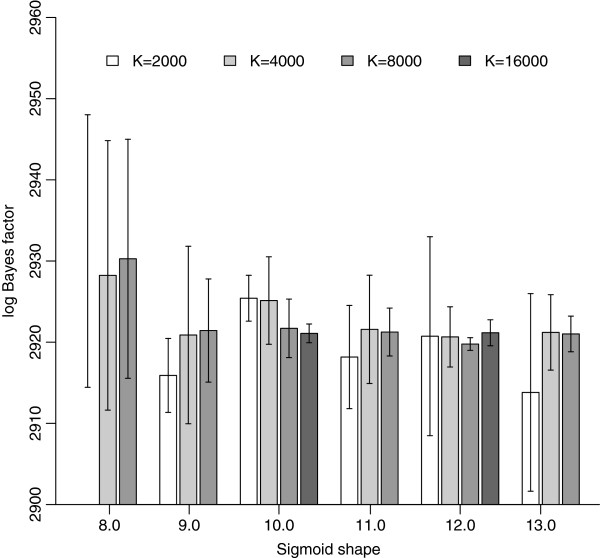
Results: We here assess the original 'model-switch' path sampling approach for direct Bayes factor estimation in phylogenetics, as well as an extension that uses more samples, to construct a direct path between two competing models, thereby eliminating the need to calculate each model's marginal likelihood independently. Further, we provide a competing Bayes factor estimator using an adaptation of the recently introduced stepping-stone sampling algorithm and set out to determine appropriate settings for accurately calculating such Bayes factors, with context-dependent evolutionary models as an example. While we show that modest efforts are required to roughly identify the increase in model fit, only drastically increased computation times ensure the accuracy needed to detect more subtle details of the evolutionary process.
Conclusions: We show that our adaptation of stepping-stone sampling for direct Bayes factor calculation outperforms the original path sampling approach as well as an extension that exploits more samples. Our proposed approach for Bayes factor estimation also has preferable statistical properties over the use of individual marginal likelihood estimates for both models under comparison. Assuming a sigmoid function to determine the path between two competing models, we provide evidence that a single well-chosen sigmoid shape value requires less computational efforts in order to approximate the true value of the (log) Bayes factor compared to the original approach. We show that the (log) Bayes factors calculated using path sampling and stepping-stone sampling differ drastically from those estimated using either of the harmonic mean estimators, supporting earlier claims that the latter systematically overestimate the performance of high-dimensional models, which we show can lead to erroneous conclusions. Based on our results, we argue that highly accurate estimation of differences in model fit for high-dimensional models requires much more computational effort than suggested in recent studies on marginal likelihood estimation.
Figures


References
-
- Baele G. Context-dependent evolutionary models for non-coding sequences: an overview of several decades of research and an analysis of Laurasiatheria and Primate evolution. Evol Biol. 2012;39:61–82. doi: 10.1007/s11692-011-9139-2. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
