

A comparison of methods for differential expression analysis of RNA-seq data
- PMID: 23497356
- PMCID: PMC3608160
- DOI: 10.1186/1471-2105-14-91
A comparison of methods for differential expression analysis of RNA-seq data
Abstract
Background: Finding genes that are differentially expressed between conditions is an integral part of understanding the molecular basis of phenotypic variation. In the past decades, DNA microarrays have been used extensively to quantify the abundance of mRNA corresponding to different genes, and more recently high-throughput sequencing of cDNA (RNA-seq) has emerged as a powerful competitor. As the cost of sequencing decreases, it is conceivable that the use of RNA-seq for differential expression analysis will increase rapidly. To exploit the possibilities and address the challenges posed by this relatively new type of data, a number of software packages have been developed especially for differential expression analysis of RNA-seq data.
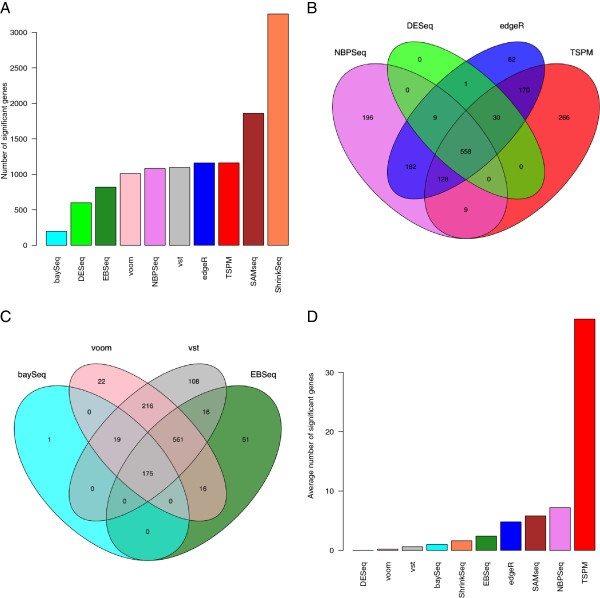
Results: We conducted an extensive comparison of eleven methods for differential expression analysis of RNA-seq data. All methods are freely available within the R framework and take as input a matrix of counts, i.e. the number of reads mapping to each genomic feature of interest in each of a number of samples. We evaluate the methods based on both simulated data and real RNA-seq data.
Conclusions: Very small sample sizes, which are still common in RNA-seq experiments, impose problems for all evaluated methods and any results obtained under such conditions should be interpreted with caution. For larger sample sizes, the methods combining a variance-stabilizing transformation with the 'limma' method for differential expression analysis perform well under many different conditions, as does the nonparametric SAMseq method.
Figures
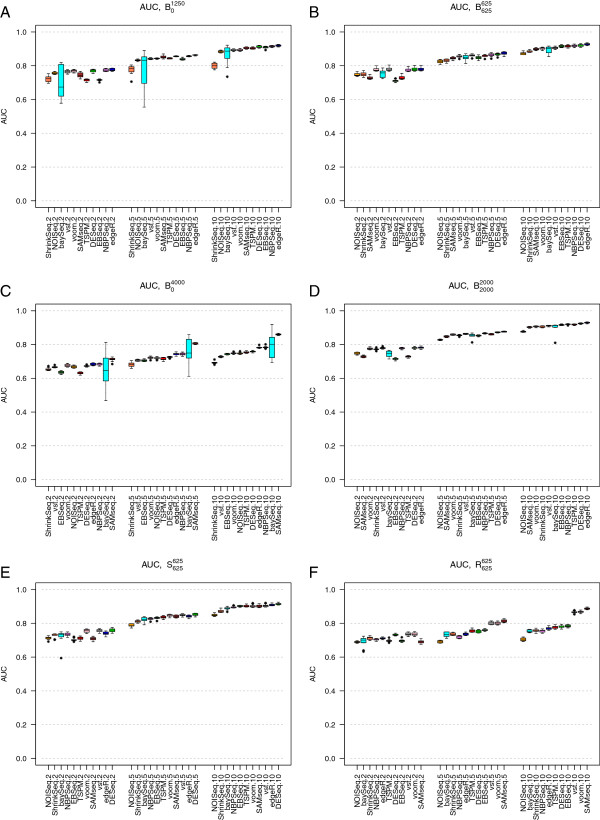
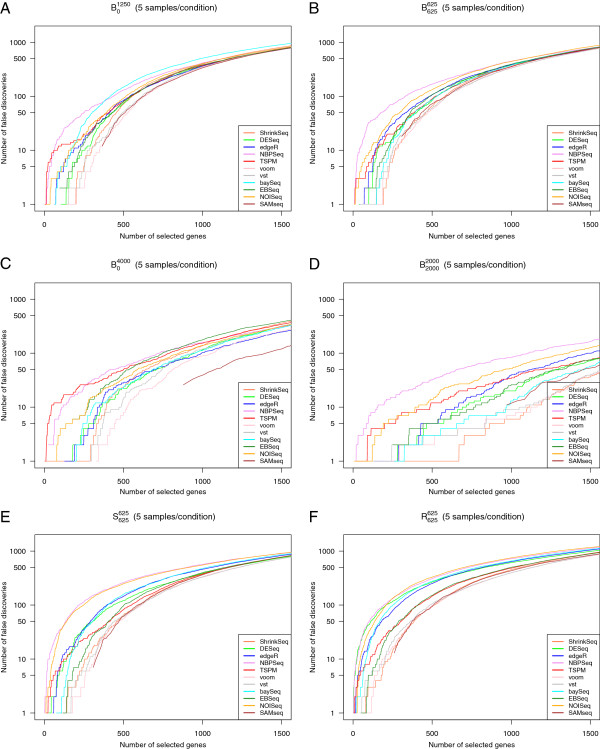
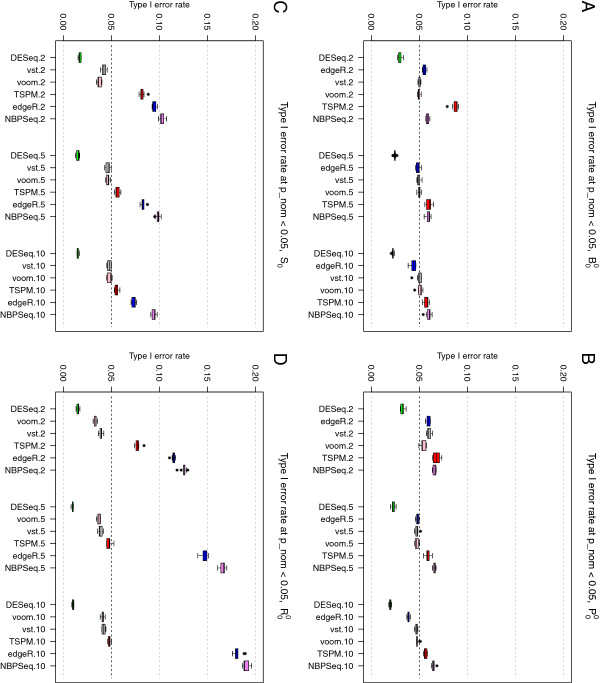
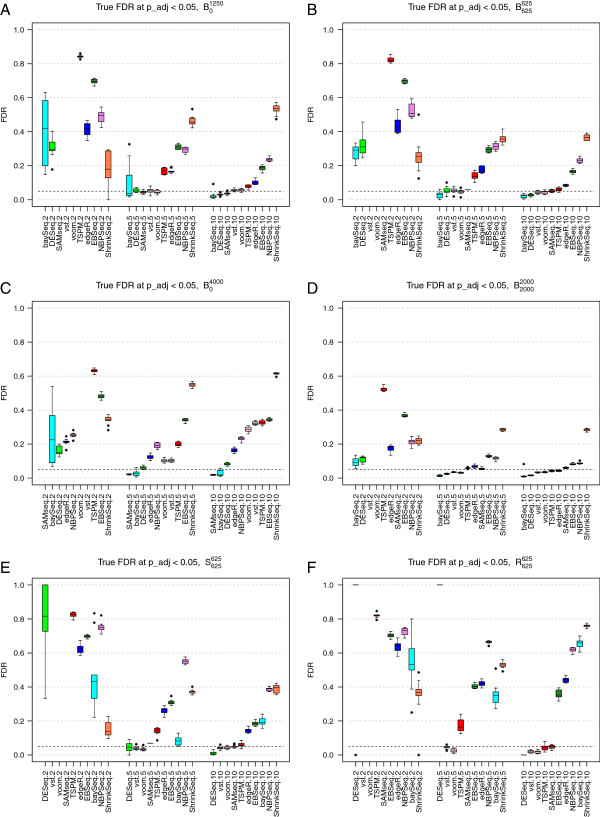
References
-
- Chen G, Wang C, Shi T. Overview of available methods for diverse RNA-Seq data analyses. Sci China Life Sci. 2011;54:1121–1128. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
