

Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum
- PMID: 23497566
- PMCID: PMC3610264
- DOI: 10.1186/1750-1172-8-41
Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum
Abstract
Background: Mutations in SACS, leading to autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), have been identified as a frequent cause of recessive early-onset ataxia around the world. Here we aimed to enlarge the spectrum of SACS mutations outside Quebec, to establish the pathogenicity of novel variants, and to expand the clinical and imaging phenotype.
Methods: Sequencing of SACS in 22 patients with unexplained early-onset ataxia, assessment of novel SACS variants in 3.500 European control chromosomes and extensive phenotypic investigations of all SACS carriers.
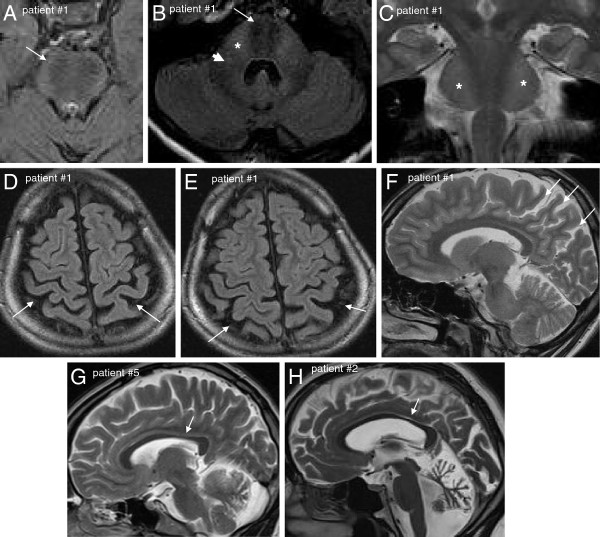
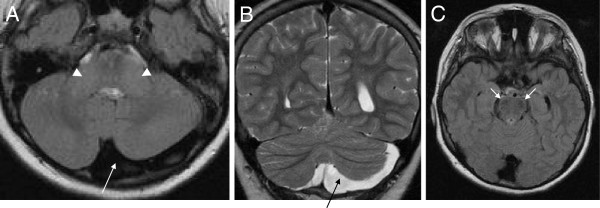
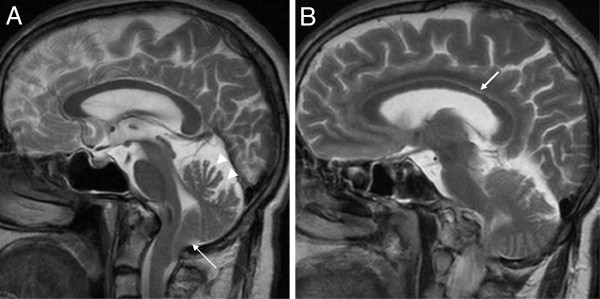
Results: We identified 11 index patients harbouring 17 novel SACS variants. 9/11 patients harboured two variants of at least probable pathogenicity which were not observed in controls and, in case of missense mutations, were located in highly conserved domains. These 9 patients accounted for at least 11% (9/83) in our series of unexplained early onset ataxia subjects. While most patients (7/9) showed the classical ARSACS triad, the presenting phenotype reached from pure neuropathy (leading to the initial diagnosis of Charcot-Marie-Tooth disease) in one subject to the absence of any signs of neuropathy in another. In contrast to its name "spastic ataxia", neither spasticity (absent in 2/9=22%) nor extensor plantar response (absent in 3/9=33%) nor cerebellar ataxia (absent in 1/9=11%) were obligate features. Autonomic features included urine urge incontinence and erectile dysfunction. Apart from the well-established MRI finding of pontine hypointensities, all patients (100%) showed hyperintensities of the lateral pons merging into the (thickened) middle cerebellar peduncles. In addition, 63% exhibited bilateral parietal cerebral atrophy, and 63% a short circumscribed thinning of the posterior midbody of the corpus callosum. In 2 further patients with differences in important clinical features, VUS class 3 variants (c.1373C>T [p.Thr458Ile] and c.2983 G>T [p.Val995Phe]) were identified. These variants were, however, also observed in controls, thus questioning their pathogenic relevance.
Conclusions: We here demonstrate that each feature of the classical ARSACS triad (cerebellar ataxia, spasticity and peripheral neuropathy) might be missing in ARSACS. Nevertheless, characteristic MRI features - which also extend to supratentorial regions and involve the cerebral cortex - will help to establish the diagnosis in most cases.
Figures

References
-
- Vermeer S, van de Warrenburg BP, Kamsteeg EJ. In: GeneReviews [Internet] Pagon RA, Bird TC, Dolan CR, Stephens K, editor. Seattle (WA): University of Washington; 2012. ARSACS. http://www.ncbi.nlm.nih.gov/books/NBK1255/; accessed November 22nd, 2012.
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
