

DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes
- PMID: 23497655
- PMCID: PMC4053839
- DOI: 10.1186/gb-2013-14-3-r21
DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes
Abstract
Background: Abnormal epigenetic marking is well documented in gene promoters of cancer cells, but the study of distal regulatory siteshas lagged behind.We performed a systematic analysis of DNA methylation sites connected with gene expression profilesacross normal and cancerous human genomes.
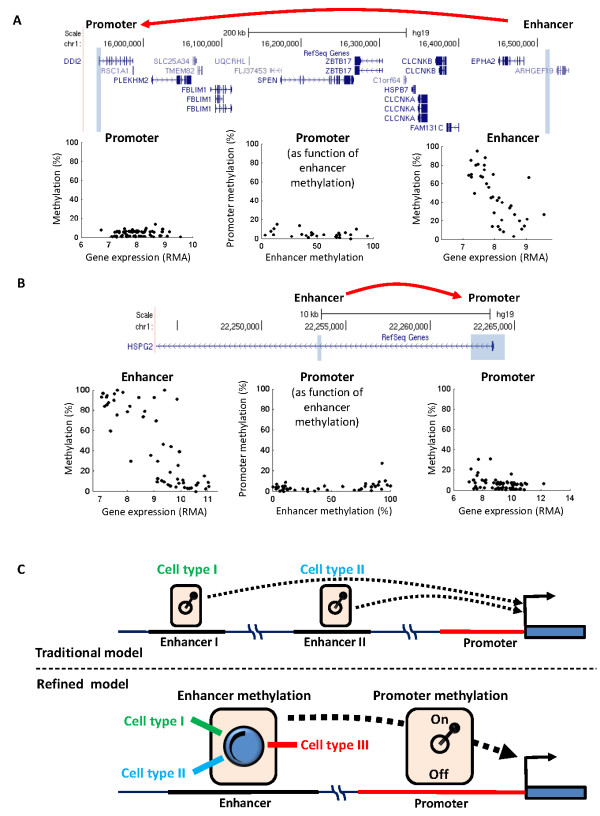
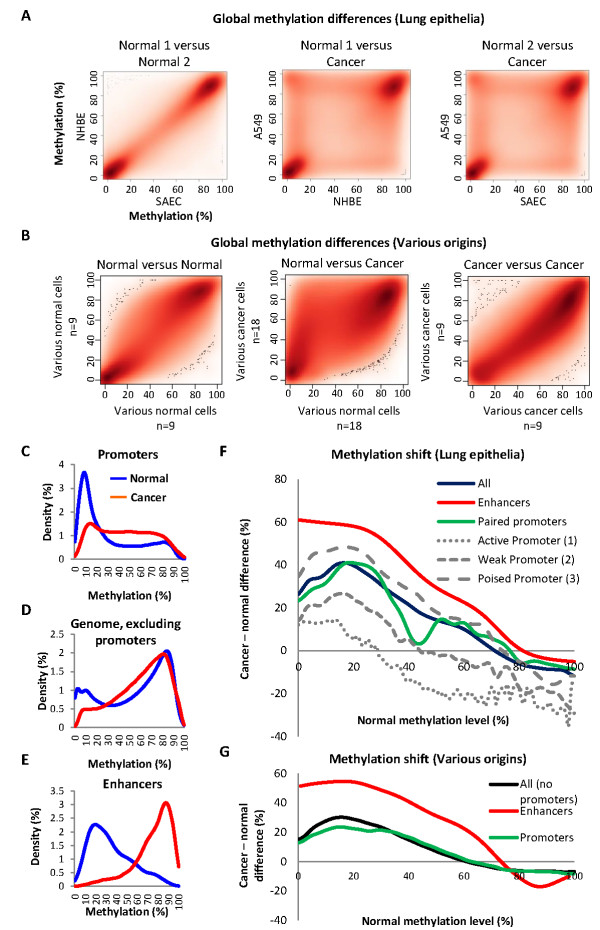
Results: Utilizing methylation and expression data in 58 cell types, we developed a model for methylation-expression relationships in gene promoters and extrapolated it to the genome. We mapped numerous sites at which DNA methylation was associated with expression of distal genes. These sites bind transcription factors in a methylation-dependent manner, and carry the chromatin marks of a particular class of transcriptional enhancers. In contrast to the traditional model of one enhancer site per cell type, we found that single enhancer sites may define gradients of expression levels across many different cell types. Strikingly, the identified sites were drastically altered in cancers: hypomethylated enhancer sites associated with upregulation of cancer-related genes and hypermethylated sites with downregulation. Moreover, the association between enhancer methylation and gene deregulation in cancerwas significantly stronger than the association of promoter methylationwith gene deregulation.
Conclusions: Methylation of distal regulatory sites is closely related to gene expression levels across the genome. Single enhancers may modulate ranges of cell-specific transcription levels, from constantlyopen promoters. In contrast to the remote relationships between promoter methylation and gene dysregulation in cancer, altered methylation of enhancer sites is closely related to gene expression profiles of transformed cells.
Figures



References
-
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;14(19):8689–8694. doi: 10.1073/pnas.1002720107. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
