

VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations
- PMID: 23498974
- PMCID: PMC3683300
- DOI: 10.1016/j.neuron.2013.02.029
VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations
Erratum in
- Neuron. 2013 Apr 24;78(2):403
Abstract
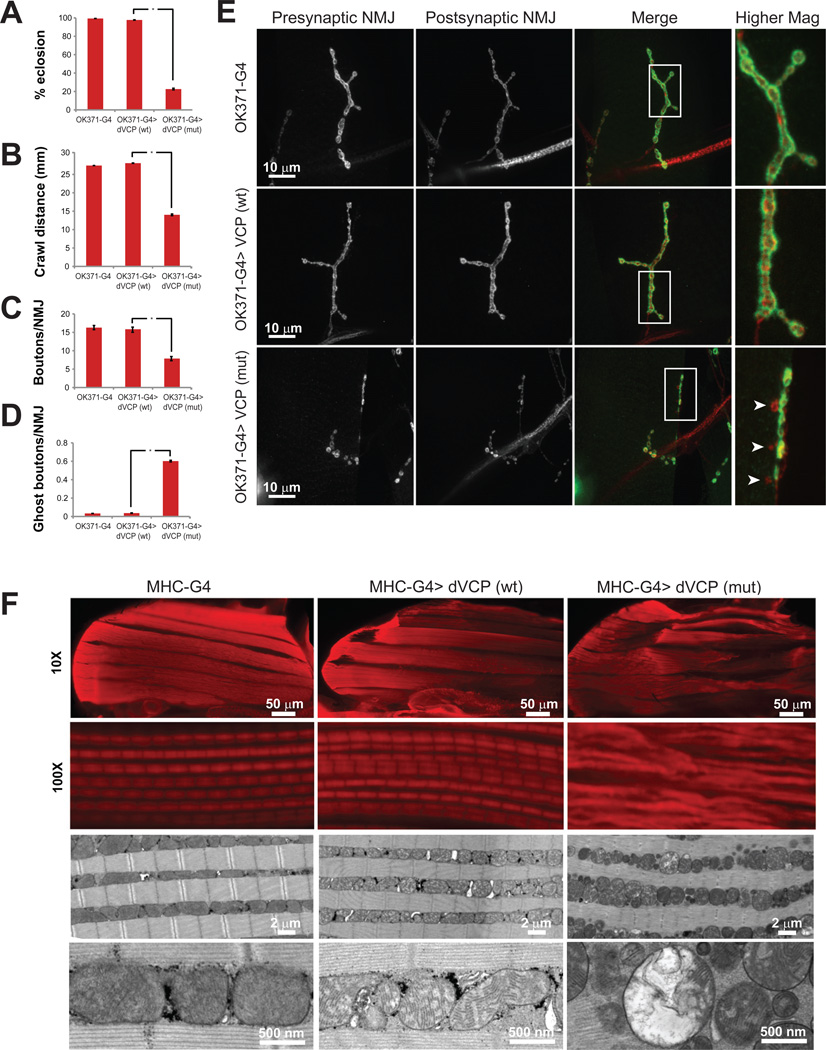
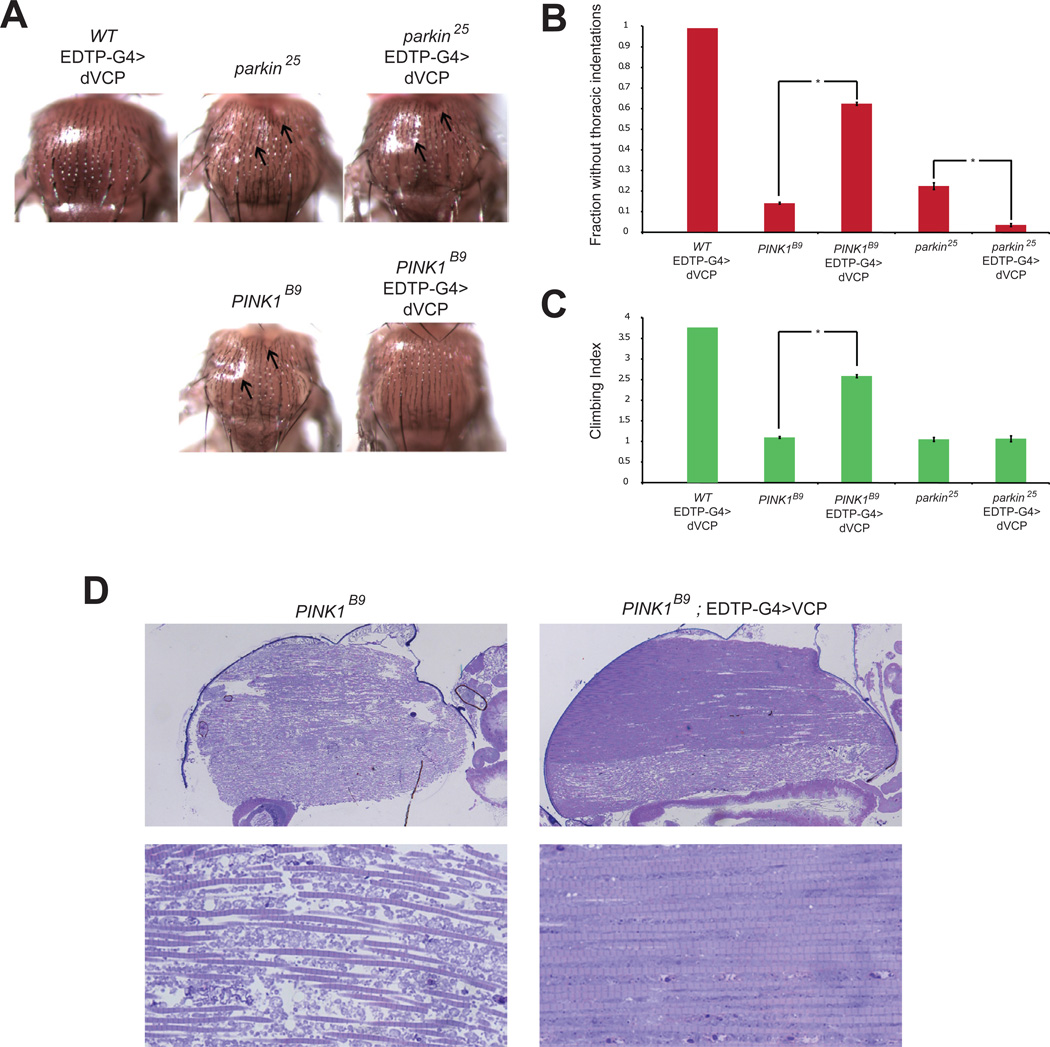
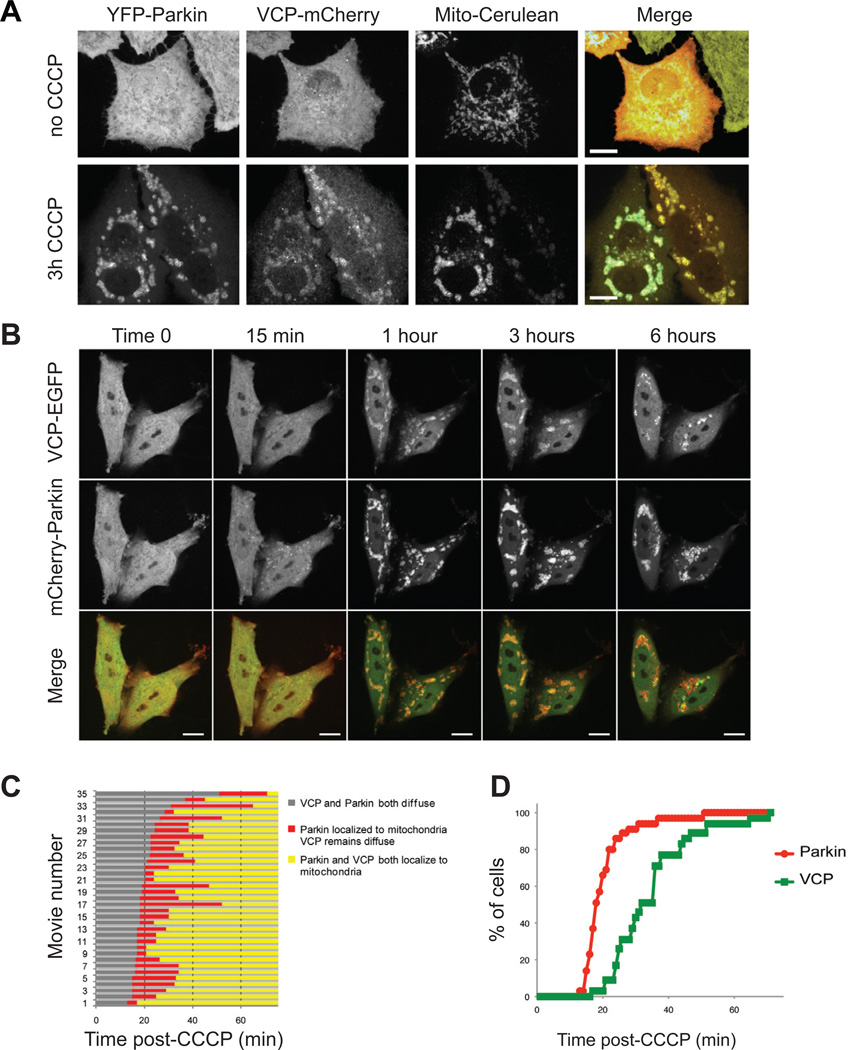
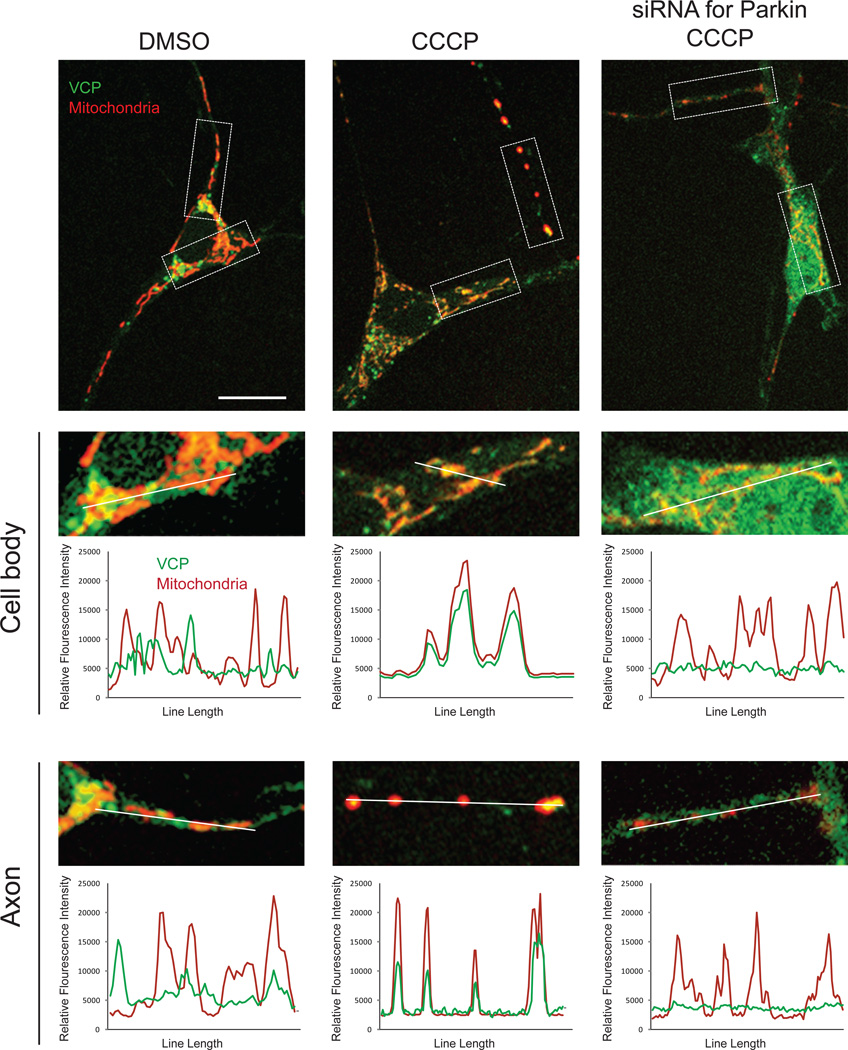
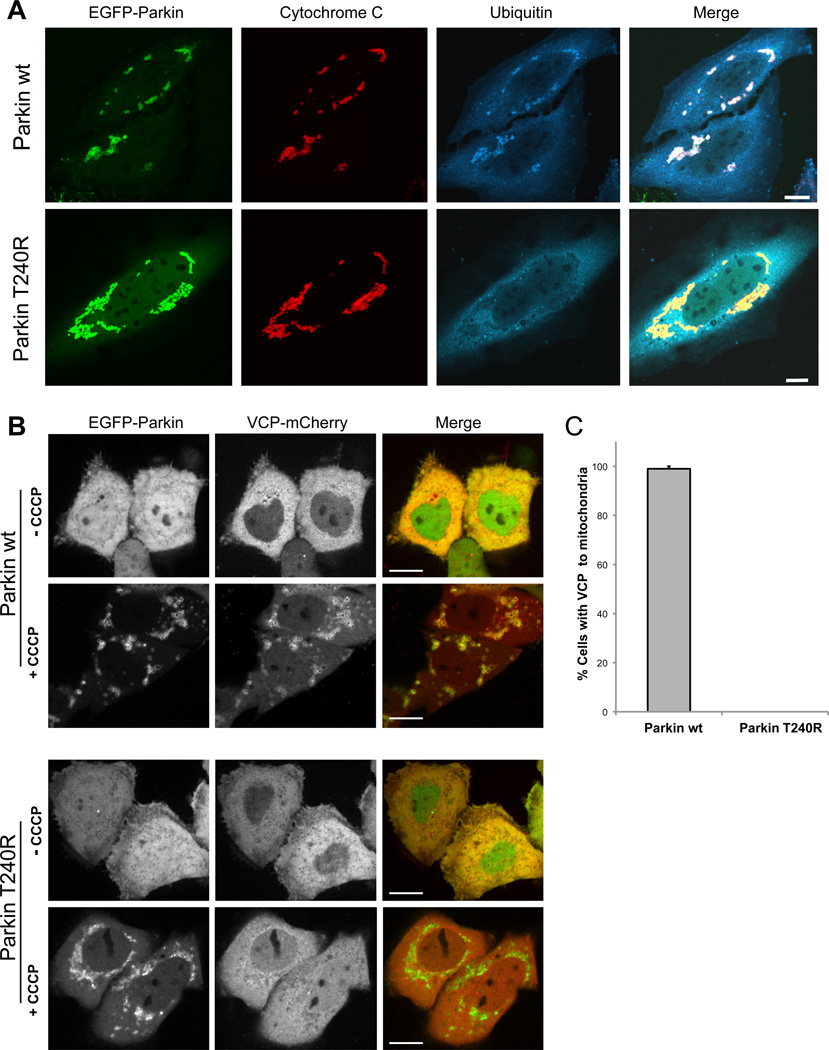
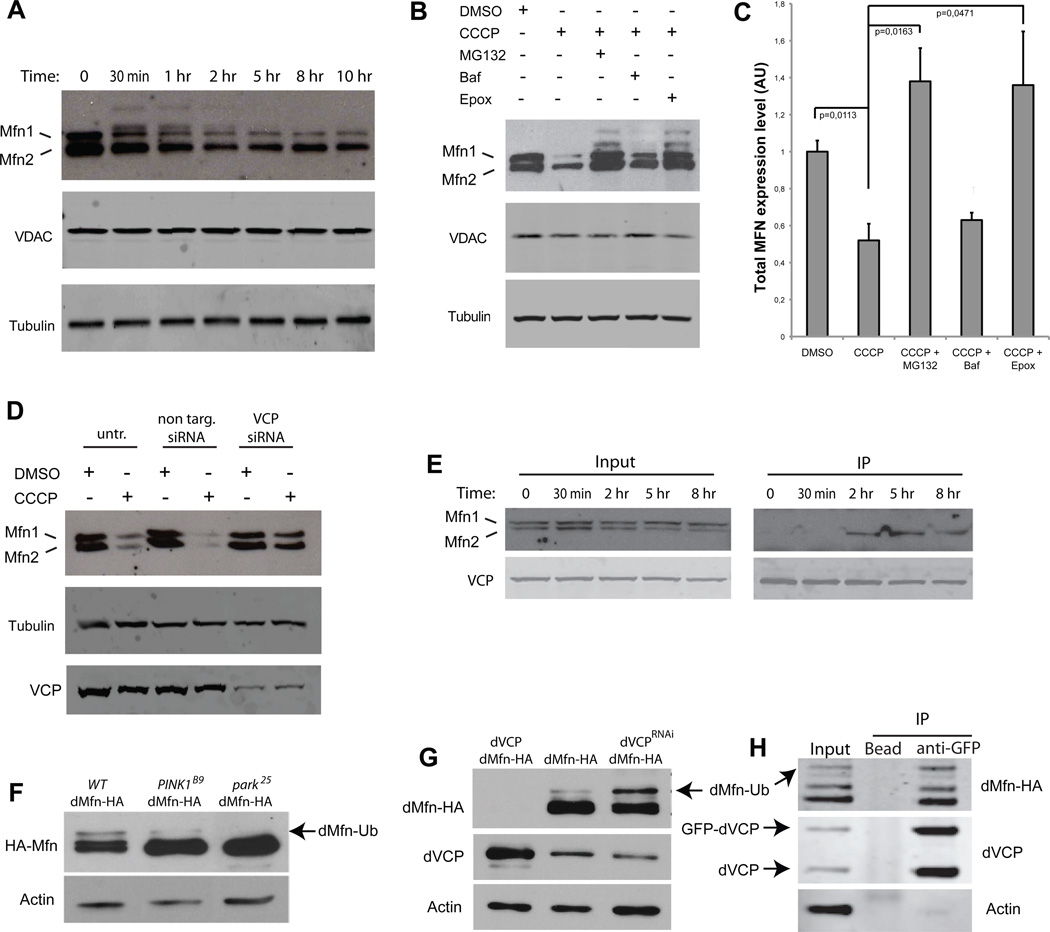
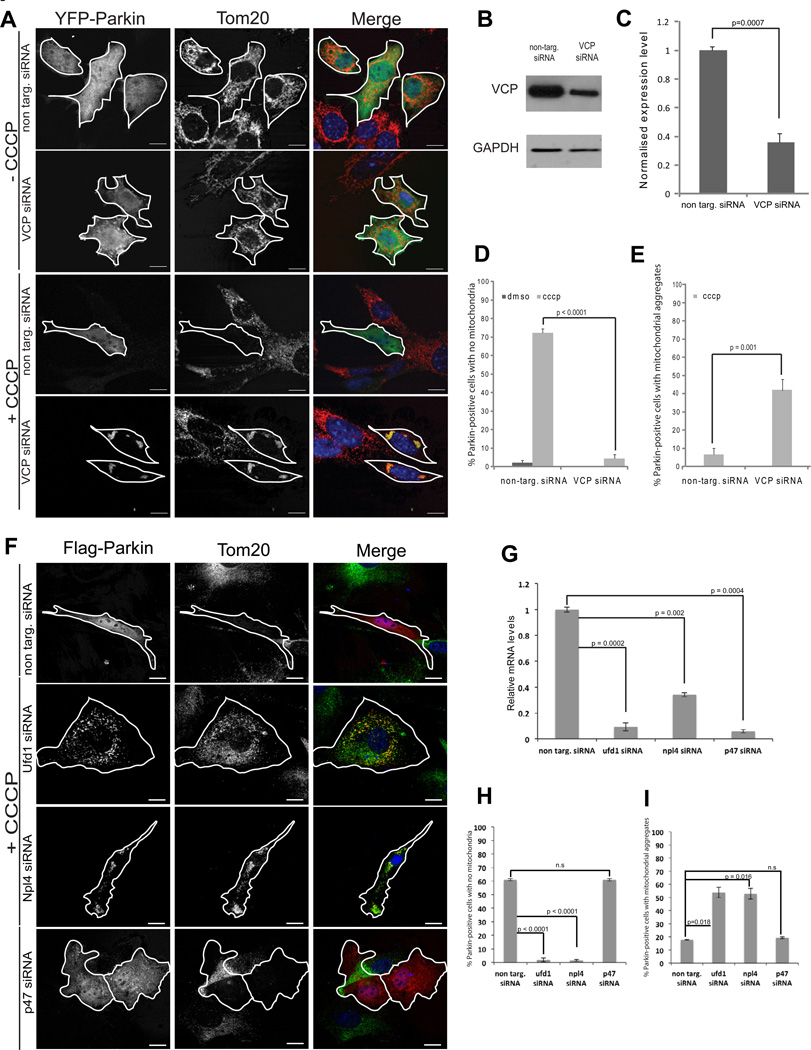
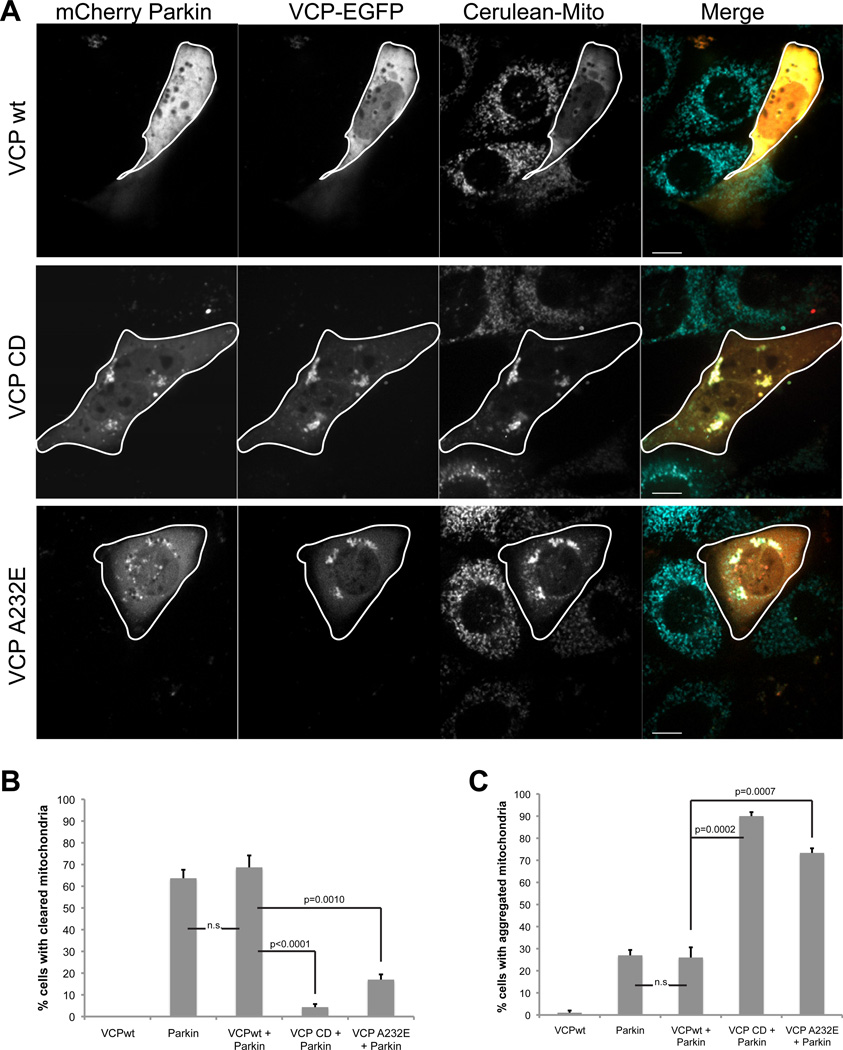
Mutations in VCP cause multisystem degeneration impacting the nervous system, muscle, and/or bone. Patients may present with ALS, Parkinsonism, frontotemporal dementia, myopathy, Paget's disease, or a combination of these. The disease mechanism is unknown. We developed a Drosophila model of VCP mutation-dependent degeneration. The phenotype is reminiscent of PINK1 and parkin mutants, including a pronounced mitochondrial defect. Indeed, VCP interacts genetically with the PINK1/parkin pathway in vivo. Paradoxically, VCP complements PINK1 deficiency but not parkin deficiency. The basis of this paradox is resolved by mechanistic studies in vitro showing that VCP recruitment to damaged mitochondria requires Parkin-mediated ubiquitination of mitochondrial targets. VCP recruitment coincides temporally with mitochondrial fission, and VCP is required for proteasome-dependent degradation of Mitofusins in vitro and in vivo. Further, VCP and its adaptor Npl4/Ufd1 are required for clearance of damaged mitochondria via the PINK1/Parkin pathway, and this is impaired by pathogenic mutations in VCP.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Neurodegenerative disease: VCP mutations lead to defects in mitochondrial dynamics.Nat Rev Neurol. 2013 May;9(5):239. doi: 10.1038/nrneurol.2013.64. Epub 2013 Apr 9. Nat Rev Neurol. 2013. PMID: 23567333 No abstract available.
References
-
- Chung PY, Beyens G, de Freitas F, Boonen S, Geusens P, Vanhoenacker F, Verbruggen L, Van Offel J, Goemaere S, Zmierczak HG, et al. Indications for a genetic association of a VCP polymorphism with the pathogenesis of sporadic Paget's disease of bone, but not for TNFSF11 (RANKL) and IL-6 polymorphisms. Molecular genetics and metabolism. 2011;103:287–292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
