

Mutations in WNT1 cause different forms of bone fragility
- PMID: 23499309
- PMCID: PMC3617378
- DOI: 10.1016/j.ajhg.2013.02.010
Mutations in WNT1 cause different forms of bone fragility
Abstract
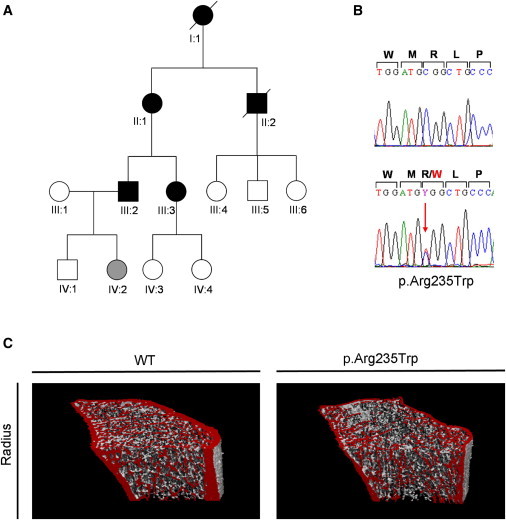
We report that hypofunctional alleles of WNT1 cause autosomal-recessive osteogenesis imperfecta, a congenital disorder characterized by reduced bone mass and recurrent fractures. In consanguineous families, we identified five homozygous mutations in WNT1: one frameshift mutation, two missense mutations, one splice-site mutation, and one nonsense mutation. In addition, in a family affected by dominantly inherited early-onset osteoporosis, a heterozygous WNT1 missense mutation was identified in affected individuals. Initial functional analysis revealed that altered WNT1 proteins fail to activate canonical LRP5-mediated WNT-regulated β-catenin signaling. Furthermore, osteoblasts cultured in vitro showed enhanced Wnt1 expression with advancing differentiation, indicating a role of WNT1 in osteoblast function and bone development. Our finding that homozygous and heterozygous variants in WNT1 predispose to low-bone-mass phenotypes might advance the development of more effective therapeutic strategies for congenital forms of bone fragility, as well as for common forms of age-related osteoporosis.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures





References
-
- Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S., Hyland J.C., Körkkö J., Prockop D.J., De Paepe A. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. - PMC - PubMed
-
- Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. - PubMed
-
- Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
