

Heme levels are increased in human failing hearts
- PMID: 23500306
- PMCID: PMC3739715
- DOI: 10.1016/j.jacc.2013.02.012
Heme levels are increased in human failing hearts
Abstract
Objectives: The goal of this study was to characterize the regulation of heme and non-heme iron in human failing hearts.
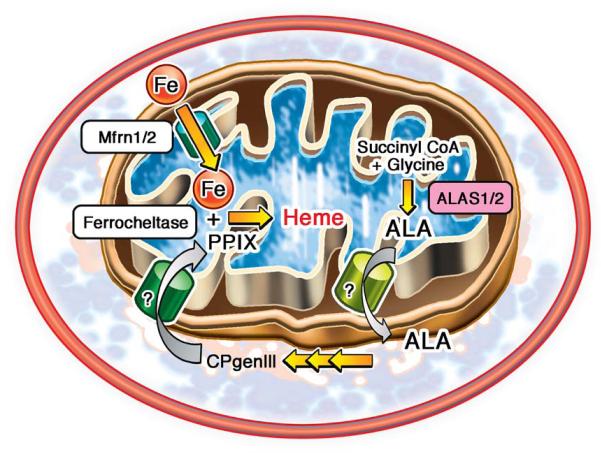
Background: Iron is an essential molecule for cellular physiology, but in excess it facilitates oxidative stress. Mitochondria are the key regulators of iron homeostasis through heme and iron-sulfur cluster synthesis. Because mitochondrial function is depressed in failing hearts and iron accumulation can lead to oxidative stress, we hypothesized that iron regulation may also be impaired in heart failure (HF).
Methods: We measured mitochondrial and cytosolic heme and non-heme iron levels in failing human hearts retrieved during cardiac transplantation surgery. In addition, we examined the expression of genes regulating cellular iron homeostasis, the heme biosynthetic pathway, and micro-RNAs that may potentially target iron regulatory networks.
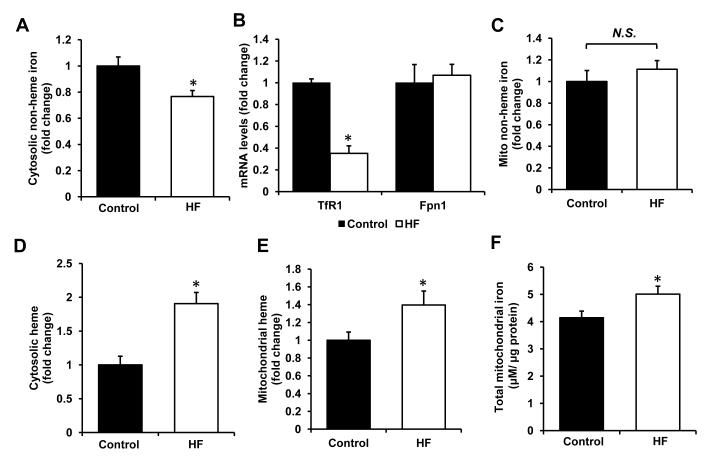
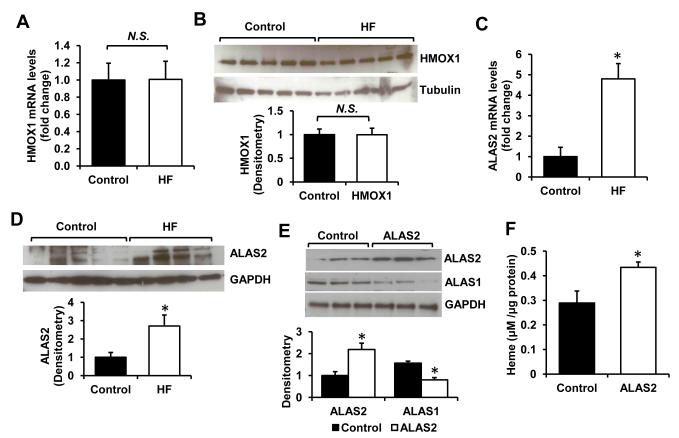
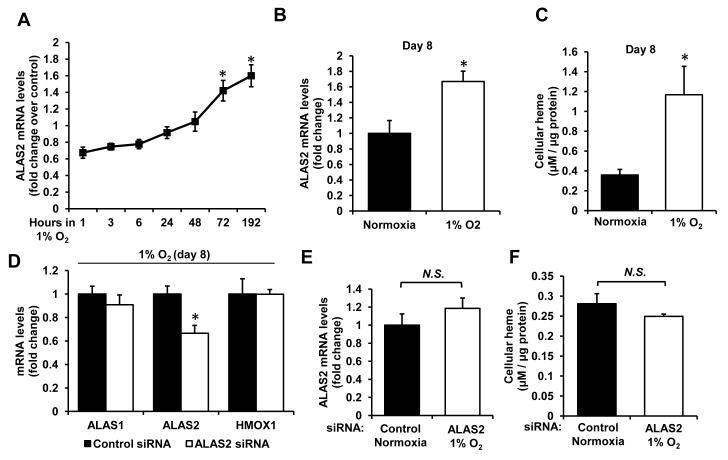
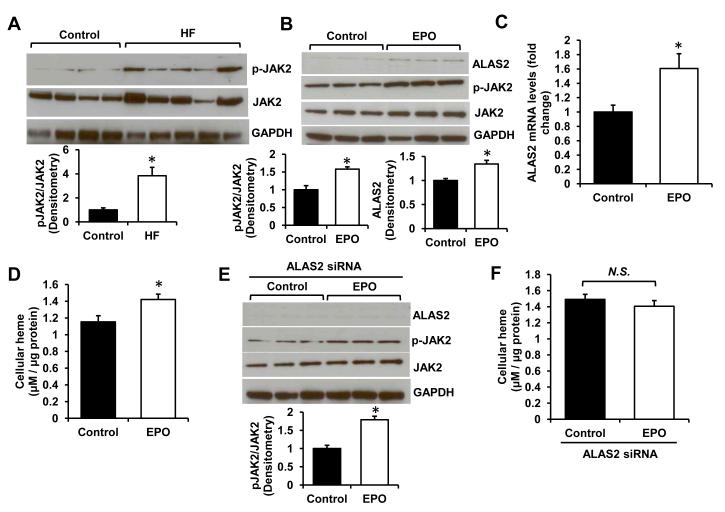
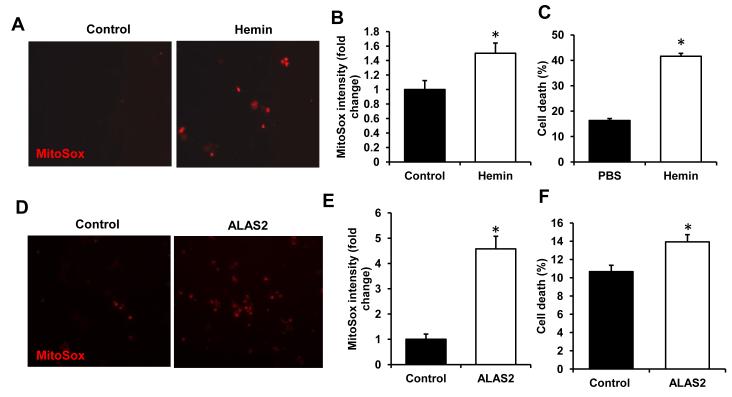
Results: Although cytosolic non-heme iron levels were reduced in HF, mitochondrial iron content was maintained. Moreover, we observed a significant increase in heme levels in failing hearts, with corresponding feedback inhibition of the heme synthetic enzymes and no change in heme degradation. The rate-limiting enzyme in heme synthesis, delta-aminolevulinic acid synthase 2 (ALAS2), was significantly upregulated in HF. Overexpression of ALAS2 in H9c2 cardiac myoblasts resulted in increased heme levels, and hypoxia and erythropoietin treatment increased heme production through upregulation of ALAS2. Finally, increased heme levels in cardiac myoblasts were associated with excess production of reactive oxygen species and cell death, suggesting a maladaptive role for increased heme in HF.
Conclusions: Despite global mitochondrial dysfunction, heme levels are maintained above baseline in human failing hearts.
Copyright © 2013 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
