

Atomic-level description of ubiquitin folding
- PMID: 23503848
- PMCID: PMC3625349
- DOI: 10.1073/pnas.1218321110
Atomic-level description of ubiquitin folding
Abstract
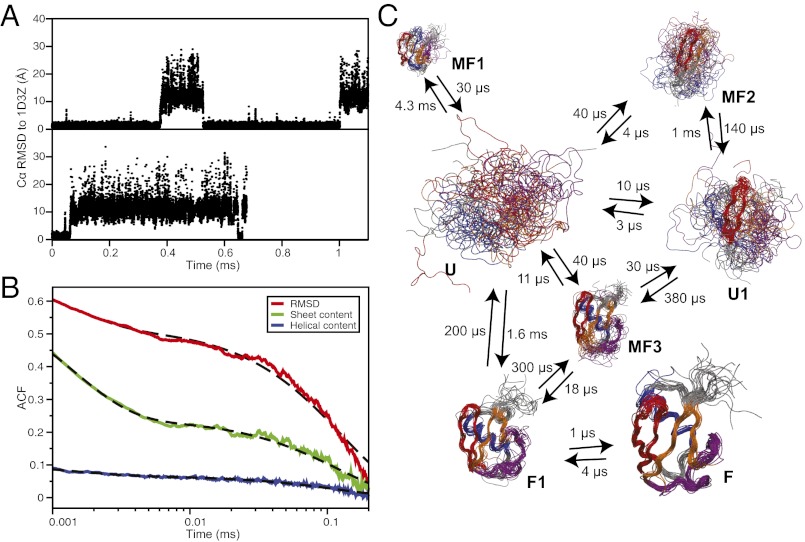
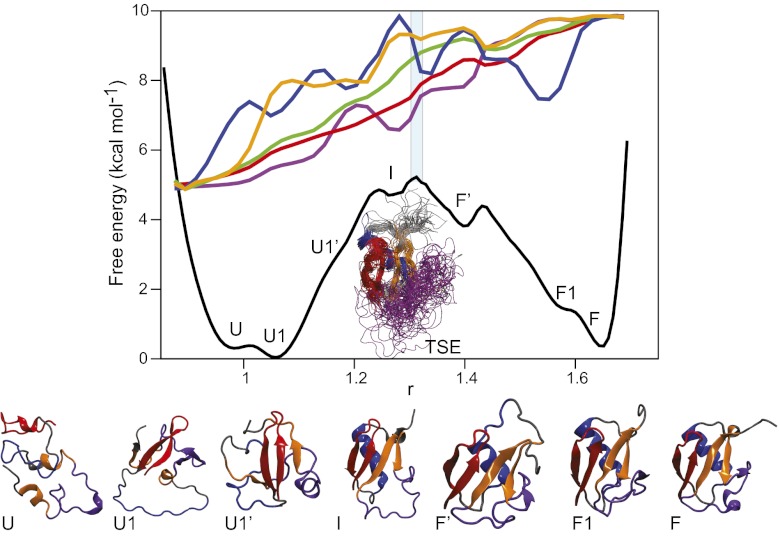
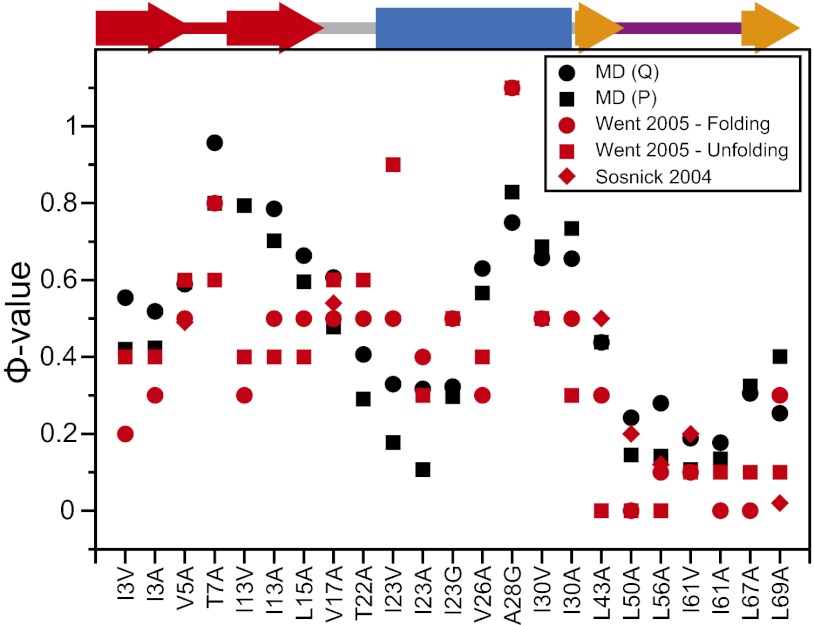
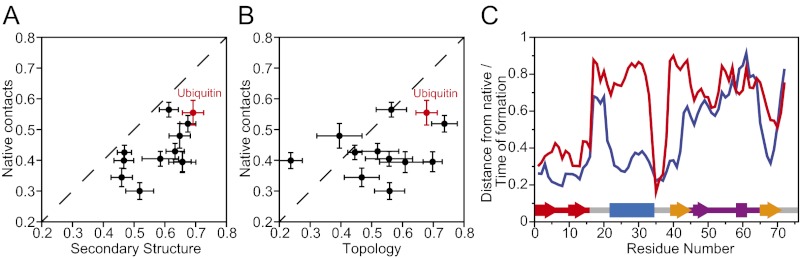
Equilibrium molecular dynamics simulations, in which proteins spontaneously and repeatedly fold and unfold, have recently been used to help elucidate the mechanistic principles that underlie the folding of fast-folding proteins. The extent to which the conclusions drawn from the analysis of such proteins, which fold on the microsecond timescale, apply to the millisecond or slower folding of naturally occurring proteins is, however, unclear. As a first attempt to address this outstanding issue, we examine here the folding of ubiquitin, a 76-residue-long protein found in all eukaryotes that is known experimentally to fold on a millisecond timescale. Ubiquitin folding has been the subject of many experimental studies, but its slow folding rate has made it difficult to observe and characterize the folding process through all-atom molecular dynamics simulations. Here we determine the mechanism, thermodynamics, and kinetics of ubiquitin folding through equilibrium atomistic simulations. The picture emerging from the simulations is in agreement with a view of ubiquitin folding suggested from previous experiments. Our findings related to the folding of ubiquitin are also consistent, for the most part, with the folding principles derived from the simulation of fast-folding proteins, suggesting that these principles may be applicable to a wider range of proteins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
A "slow" protein folds quickly in the end.Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5744-5. doi: 10.1073/pnas.1303539110. Epub 2013 Apr 1. Proc Natl Acad Sci U S A. 2013. PMID: 23572570 Free PMC article. No abstract available.
References
-
- Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14(1):76–88. - PubMed
-
- Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. Sub-microsecond protein folding. J Mol Biol. 2006;359(3):546–553. - PubMed
-
- Horng JC, Moroz V, Raleigh DP. Rapid cooperative two-state folding of a miniature alpha-beta protein and design of a thermostable variant. J Mol Biol. 2003;326(4):1261–1270. - PubMed
-
- Nauli S, Kuhlman B, Baker D. Computer-based redesign of a protein folding pathway. Nat Struct Biol. 2001;8(7):602–605. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
