

Dissection of regulatory networks that are altered in disease via differential co-expression
- PMID: 23505361
- PMCID: PMC3591264
- DOI: 10.1371/journal.pcbi.1002955
Dissection of regulatory networks that are altered in disease via differential co-expression
Abstract
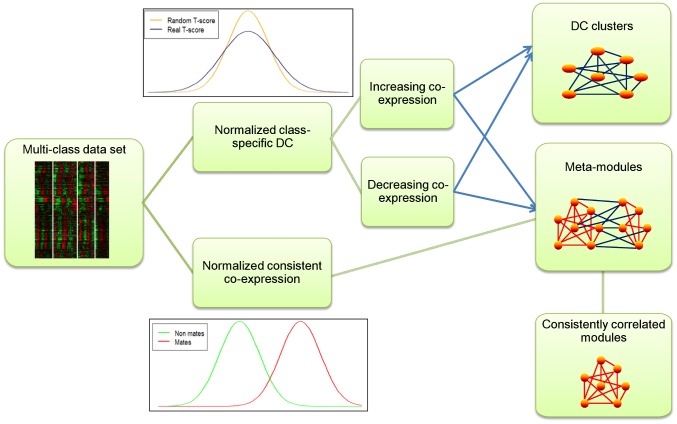
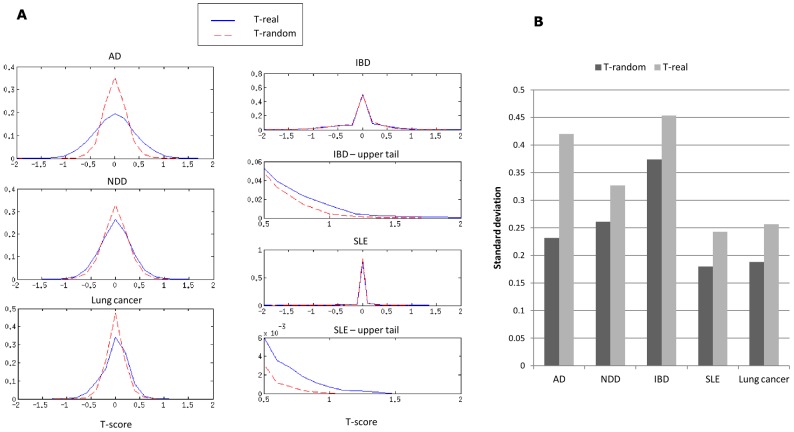
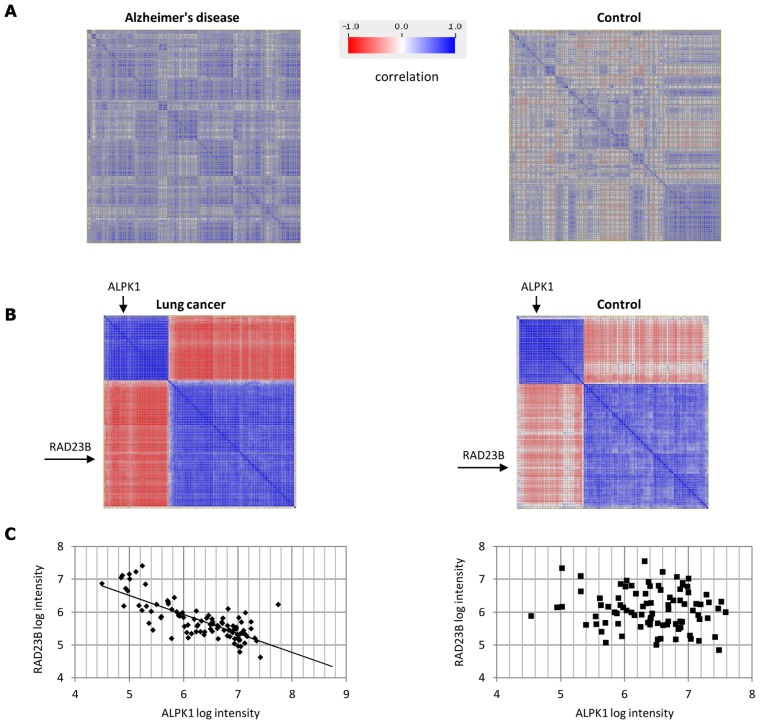
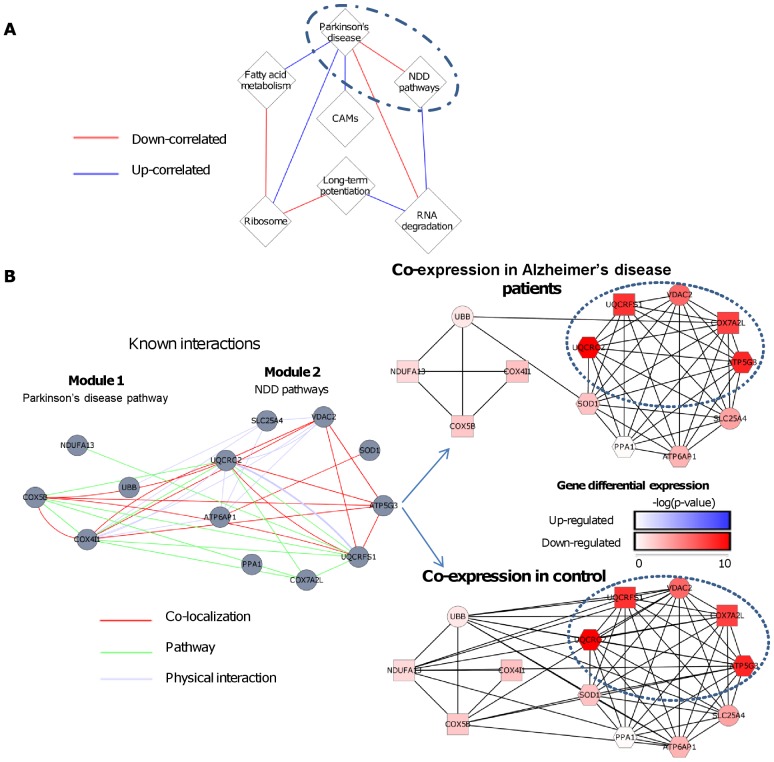
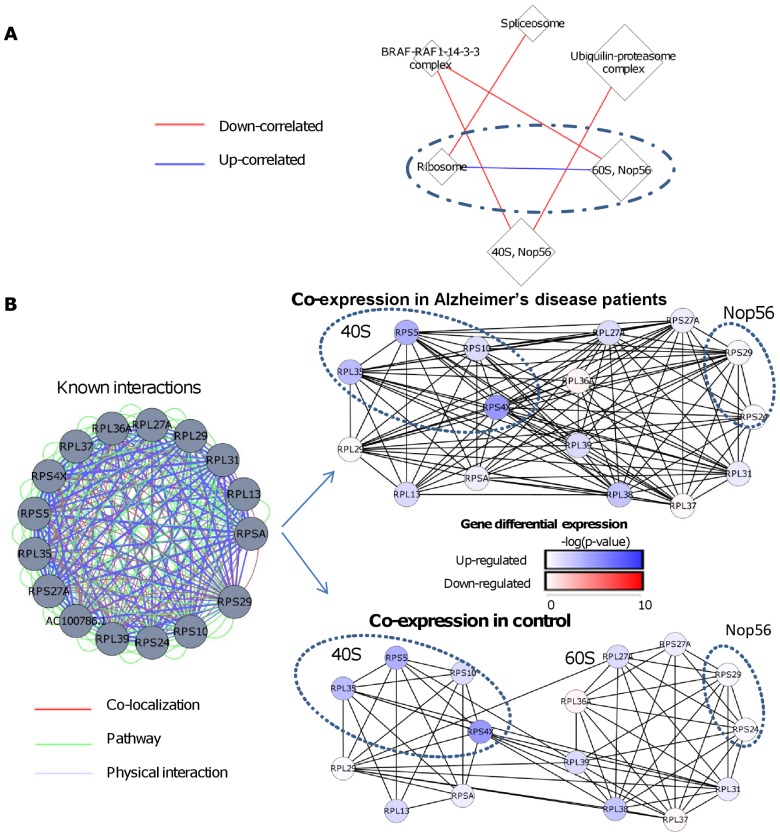
Comparing the gene-expression profiles of sick and healthy individuals can help in understanding disease. Such differential expression analysis is a well-established way to find gene sets whose expression is altered in the disease. Recent approaches to gene-expression analysis go a step further and seek differential co-expression patterns, wherein the level of co-expression of a set of genes differs markedly between disease and control samples. Such patterns can arise from a disease-related change in the regulatory mechanism governing that set of genes, and pinpoint dysfunctional regulatory networks. Here we present DICER, a new method for detecting differentially co-expressed gene sets using a novel probabilistic score for differential correlation. DICER goes beyond standard differential co-expression and detects pairs of modules showing differential co-expression. The expression profiles of genes within each module of the pair are correlated across all samples. The correlation between the two modules, however, differs markedly between the disease and normal samples. We show that DICER outperforms the state of the art in terms of significance and interpretability of the detected gene sets. Moreover, the gene sets discovered by DICER manifest regulation by disease-specific microRNA families. In a case study on Alzheimer's disease, DICER dissected biological processes and protein complexes into functional subunits that are differentially co-expressed, thereby revealing inner structures in disease regulatory networks.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
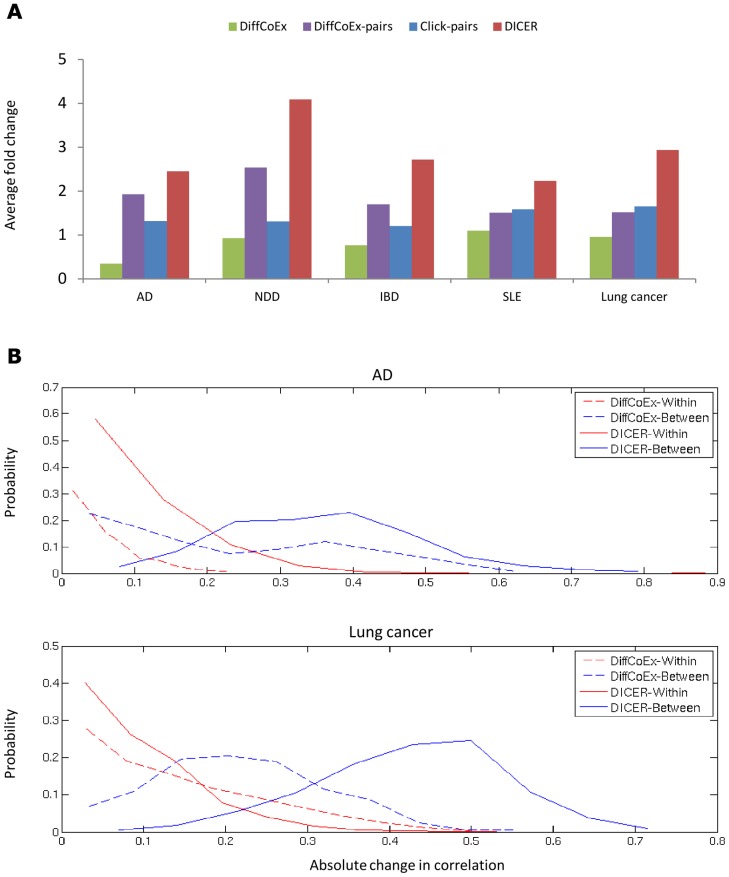
 15 genes. For DICER (red bars), the top ten up-correlated and the top ten down-correlated module pairs were taken into account. (B) The distribution of within- and between-module absolute change in correlation for DICER and DiffCoEx in the AD and lung cancer data sets.
15 genes. For DICER (red bars), the top ten up-correlated and the top ten down-correlated module pairs were taken into account. (B) The distribution of within- and between-module absolute change in correlation for DICER and DiffCoEx in the AD and lung cancer data sets.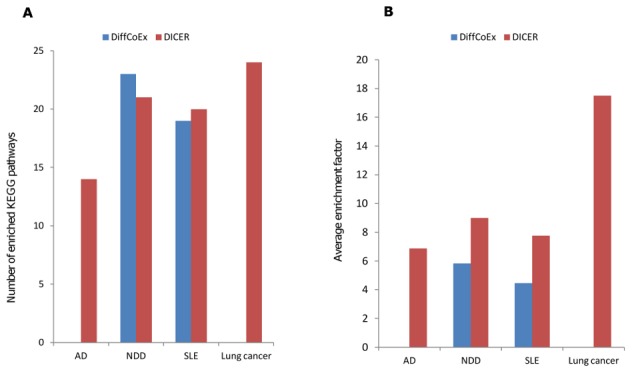
References
-
- Schulze A, Downward J (2001) Navigating gene expression using microarrays - a technology review. Nature Cell Biology 3: E190–E195. - PubMed
-
- Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5: 621–628. - PubMed
-
- Cloonan N, Forrest ARR, Kolle G, Gardiner BBA, Faulkner GJ, et al. (2008) Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nature Methods 5: 613–619. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
