

Adaptive filtering of microarray gene expression data based on Gaussian mixture decomposition
- PMID: 23510016
- PMCID: PMC3637832
- DOI: 10.1186/1471-2105-14-101
Adaptive filtering of microarray gene expression data based on Gaussian mixture decomposition
Abstract
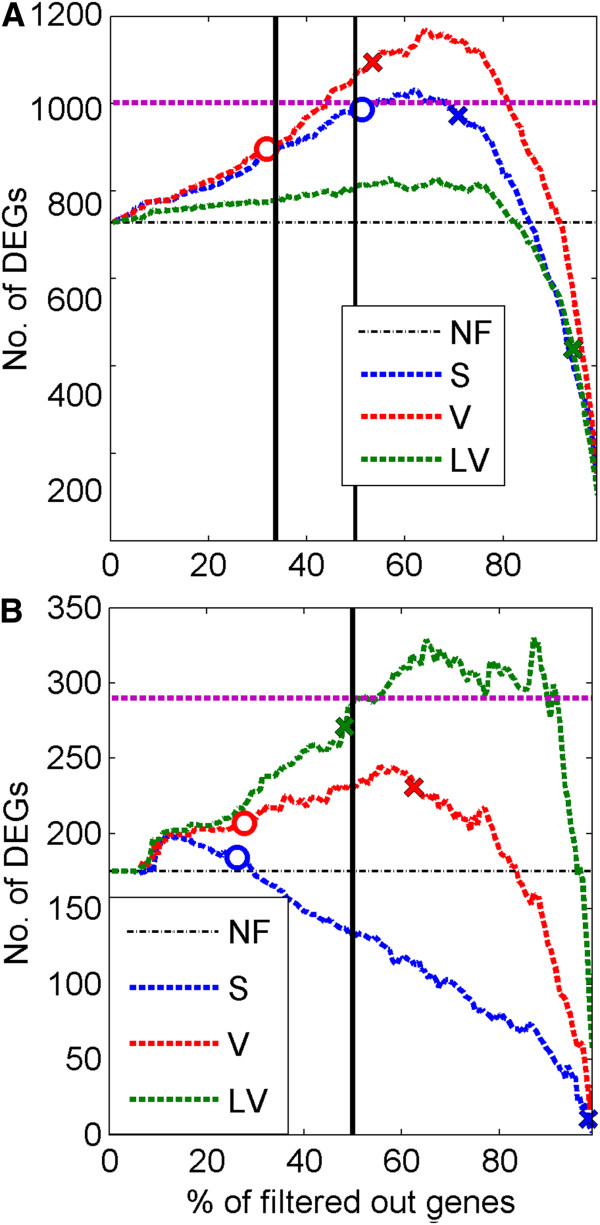
Background: DNA microarrays are used for discovery of genes expressed differentially between various biological conditions. In microarray experiments the number of analyzed samples is often much lower than the number of genes (probe sets) which leads to many false discoveries. Multiple testing correction methods control the number of false discoveries but decrease the sensitivity of discovering differentially expressed genes. Concerning this problem, filtering methods for improving the power of detection of differentially expressed genes were proposed in earlier papers. These techniques are two-step procedures, where in the first step some pool of non-informative genes is removed and in the second step only the pool of the retained genes is used for searching for differentially expressed genes.
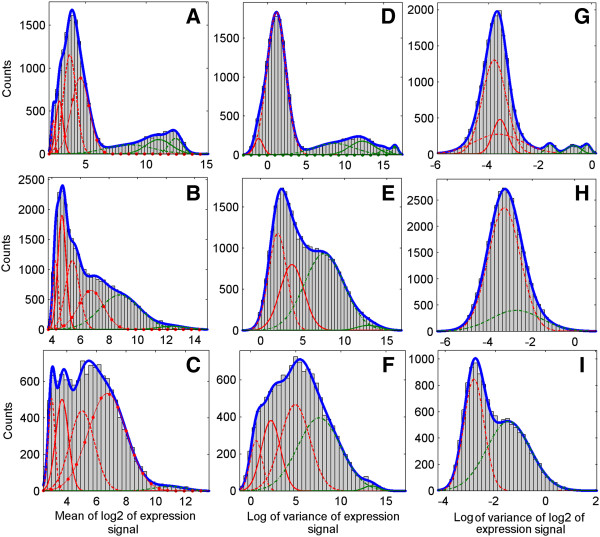
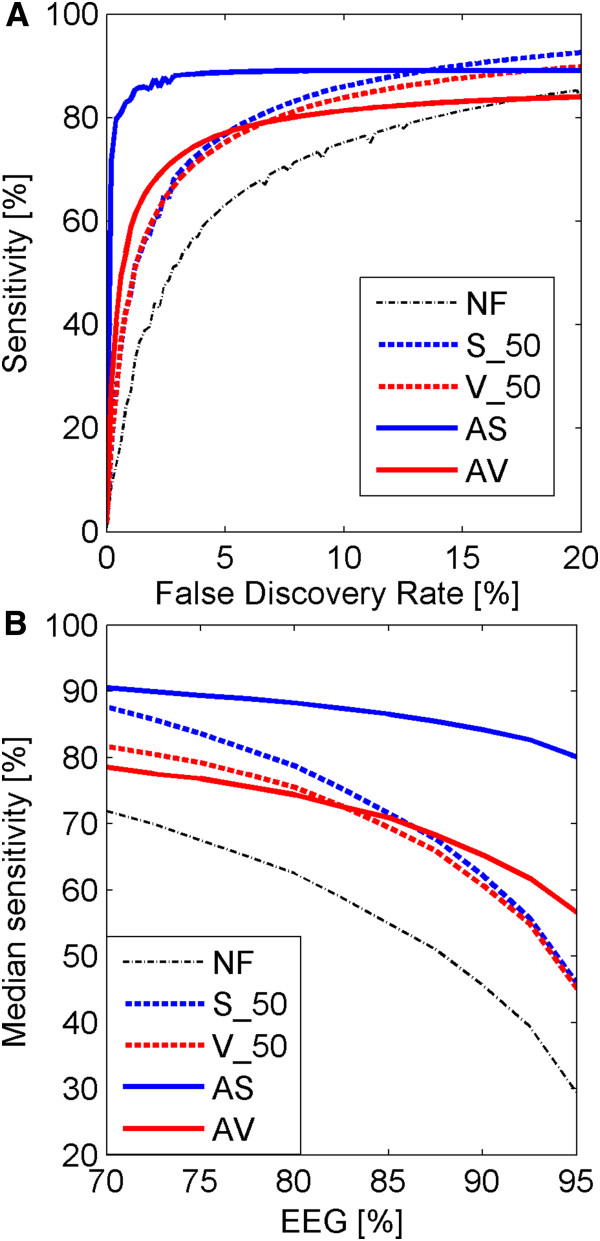
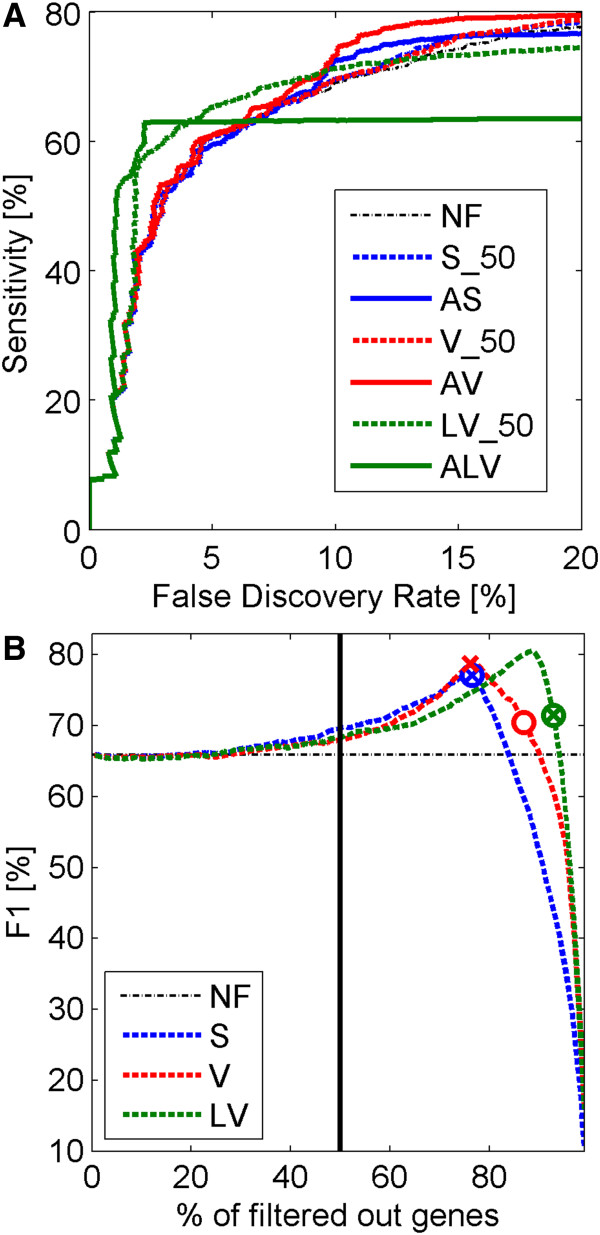
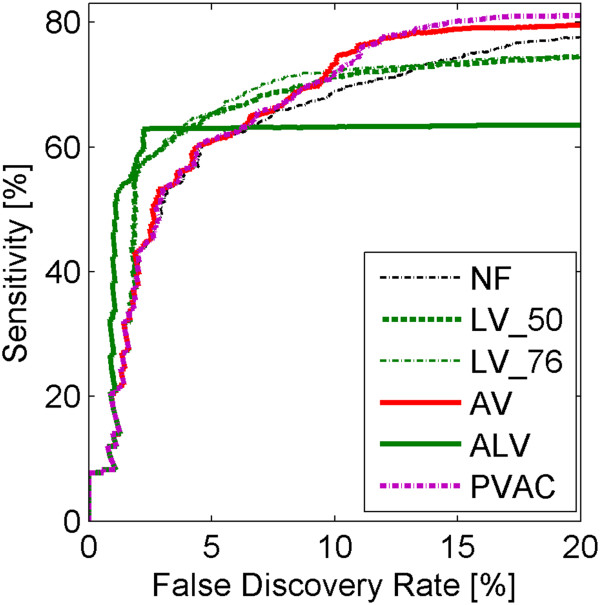
Results: A very important parameter to choose is the proportion between the sizes of the pools of removed and retained genes. A new method, which we propose, allow to determine close to optimal threshold values for sample means and sample variances for gene filtering. The method is adaptive and based on the decomposition of the histogram of gene expression means or variances into mixture of Gaussian components.
Conclusions: By performing analyses of several publicly available datasets and simulated datasets we demonstrate that our adaptive method increases sensitivity of finding differentially expressed genes compared to previous methods of filtering microarray data based on using fixed threshold values.
Figures
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
