

Genetics of prion diseases
- PMID: 23518043
- PMCID: PMC3705206
- DOI: 10.1016/j.gde.2013.02.012
Genetics of prion diseases
Abstract
Prion diseases are transmissible, fatal neurodegenerative diseases that include scrapie and bovine spongiform encephalopathy (BSE) in animals and Creutzfeldt-Jakob disease (CJD) in human. The prion protein gene (PRNP) is the major genetic determinant of susceptibility, however, several studies now suggest that other genes are also important. Two recent genome wide association studies in human have identified four new loci of interest: ZBTB38-RASA2 in UK CJD cases and MTMR7 and NPAS2 in variant CJD. Complementary studies in mouse have used complex crosses to identify new modifiers such as Cpne8 and provided supporting evidence for previously implicated genes (Rarb and Stmn2). Expression profiling has identified new candidates, including Hspa13, which reduces incubation time in a transgenic model.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
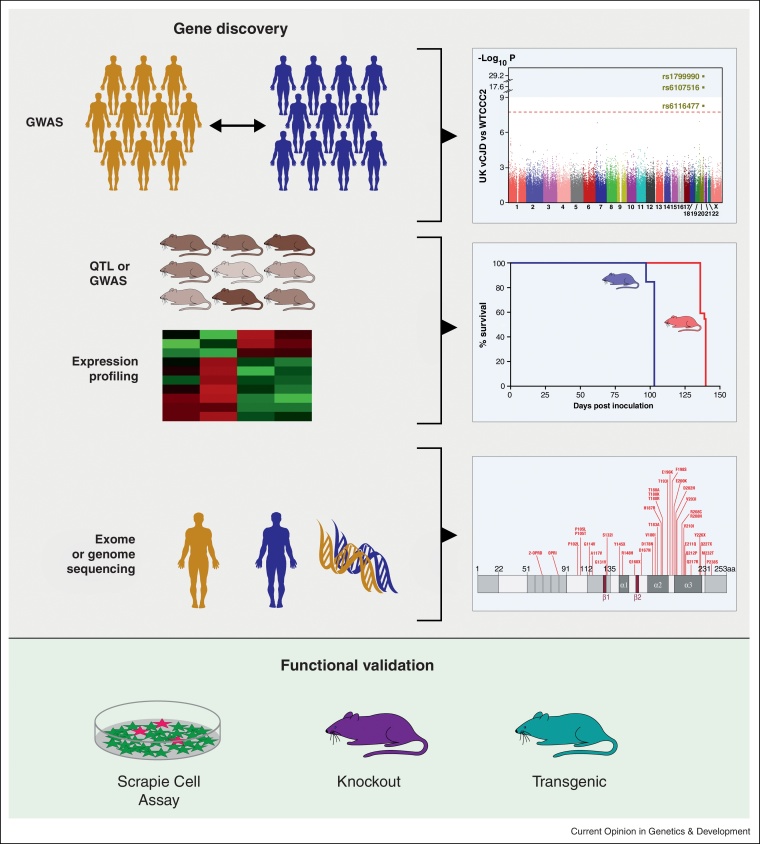
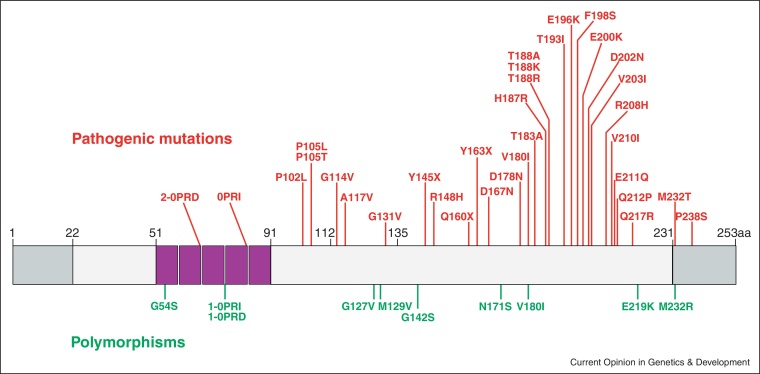
References
-
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Ann Rev Neurosci. 2001;24:519–550. - PubMed
-
- Ironside J.W. Variant Creutzfeldt–Jakob disease: an update. Folia Neuropathol. 2012;50:50–56. - PubMed
-
- Owen F., Poulter M., Lofthouse R., Collinge J., Crow T.J., Risby D., Baker H.F., Ridley R.M., Hsiao K., Prusiner S.B. Insertion in prion protein gene in familial Creutzfeldt–Jakob disease. Lancet. 1989;1:51–52. - PubMed
-
- Hsiao K., Baker H.F., Crow T.J., Poulter M., Owen F., Terwilliger J.D., Westaway D., Ott J., Prusiner S.B. Linkage of a prion protein missense variant to Gerstmann–Straussler syndrome. Nature. 1989;338:342–345. - PubMed
-
- Palmer M.S., Dryden A.J., Hughes J.T., Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature. 1991;352:340–342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
