

Hepatitis C virus-induced mitochondrial dysfunctions
- PMID: 23518579
- PMCID: PMC3705306
- DOI: 10.3390/v5030954
Hepatitis C virus-induced mitochondrial dysfunctions
Abstract
Chronic hepatitis C is characterized by metabolic disorders and a microenvironment in the liver dominated by oxidative stress, inflammation and regeneration processes that lead in the long term to hepatocellular carcinoma. Many lines of evidence suggest that mitochondrial dysfunctions, including modification of metabolic fluxes, generation and elimination of oxidative stress, Ca2+ signaling and apoptosis, play a central role in these processes. However, how these dysfunctions are induced by the virus and whether they play a role in disease progression and neoplastic transformation remains to be determined. Most in vitro studies performed so far have shown that several of the hepatitis C virus (HCV) proteins localize to mitochondria, but the consequences of these interactions on mitochondrial functions remain contradictory, probably due to the use of artificial expression and replication systems. In vivo studies are hampered by the fact that innate and adaptive immune responses will overlay mitochondrial dysfunctions induced directly in the hepatocyte by HCV. Thus, the molecular aspects underlying HCV-induced mitochondrial dysfunctions and their roles in viral replication and the associated pathology need yet to be confirmed in the context of productively replicating virus and physiologically relevant in vitro and in vivo model systems.
Figures
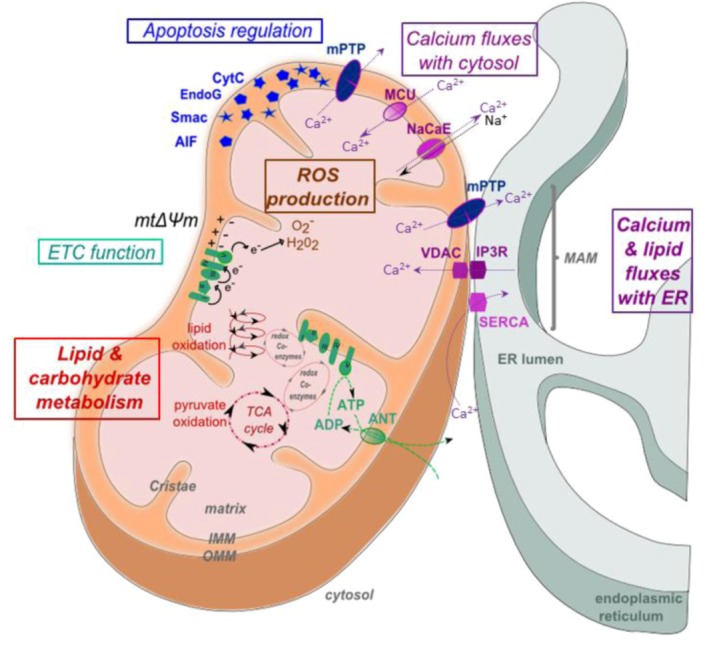
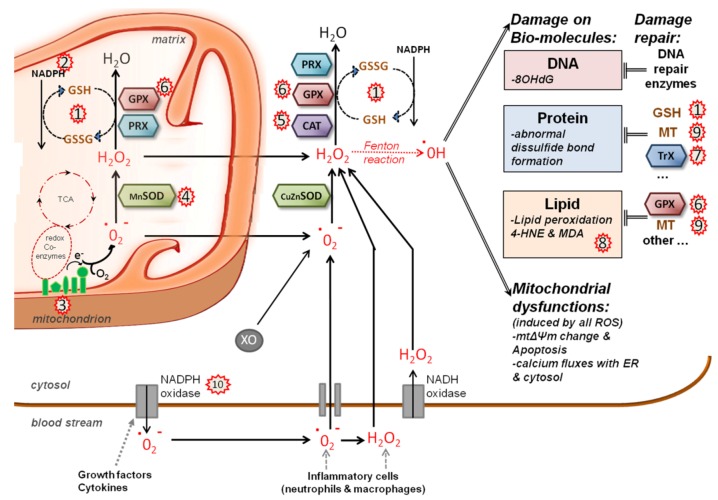
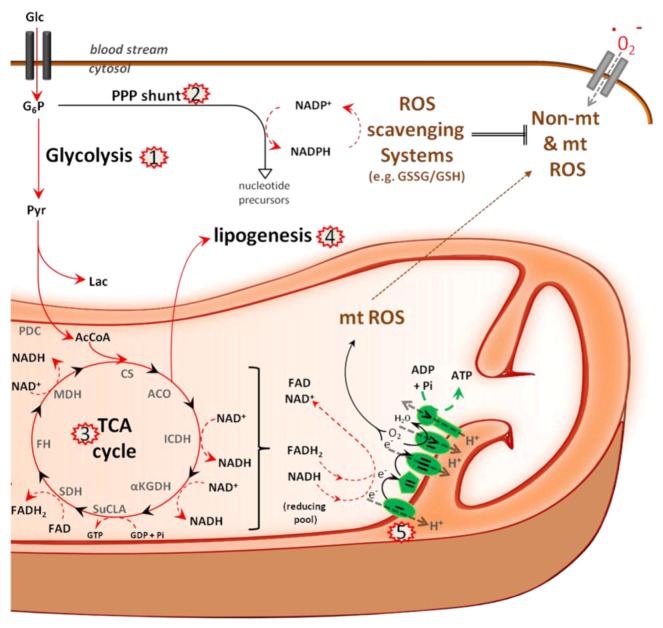
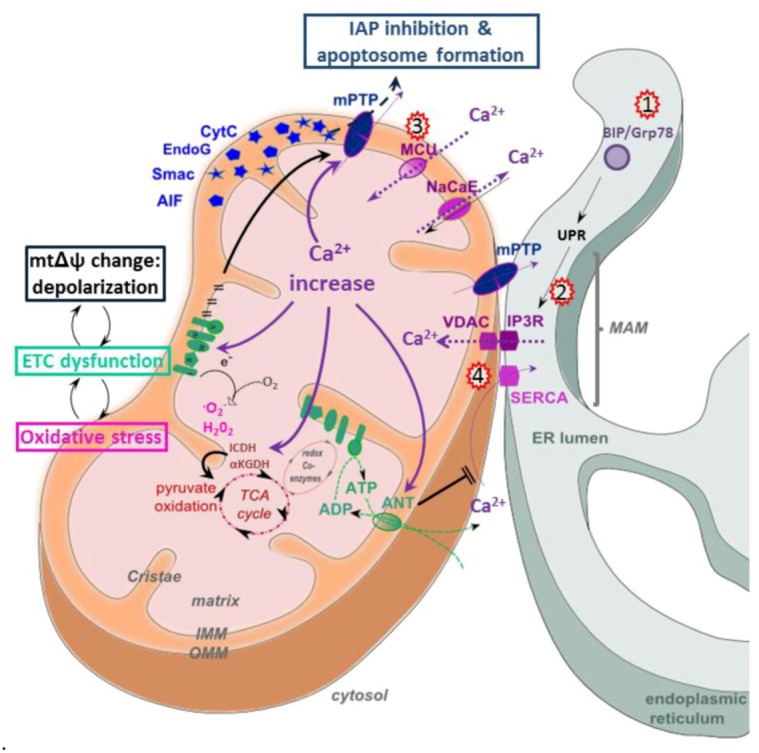
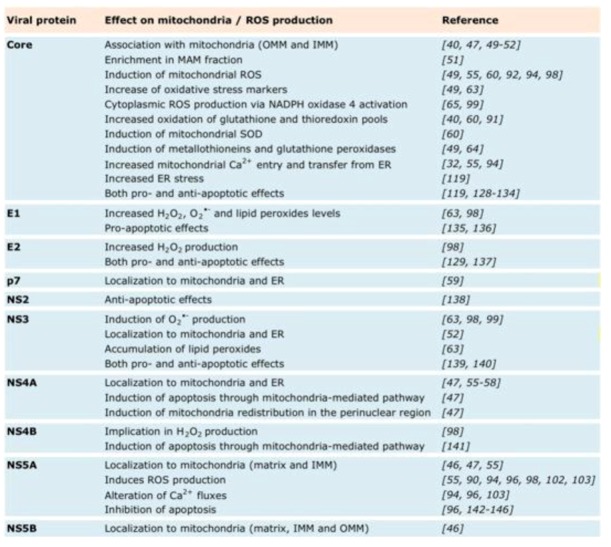
References
-
- Lemon S.M., Walker C.M., Alter M.J., Yi M.–K. Hepatitis c virus. In: Knipe D.M., Howley P.M., editors. Fields Virology. 5th. 2007. pp. 1253–1304.
-
- Ge D., Fellay J., Thompson A.J., Simon J.S., Shianna K.V., Urban T.J., Heinzen E.L., Qiu P., Bertelsen A.H., Muir A.J., et al. Genetic variation in il28b predicts hepatitis c treatment–induced viral clearance. Nature. 2009;461:399–401. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
