

PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage
- PMID: 23519076
- PMCID: PMC3679455
- DOI: 10.1038/cdd.2013.19
PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage
Abstract
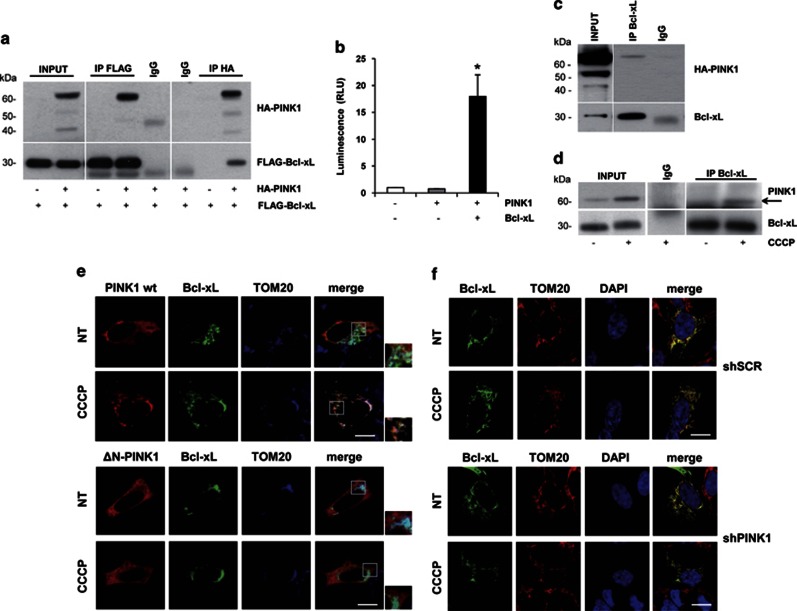
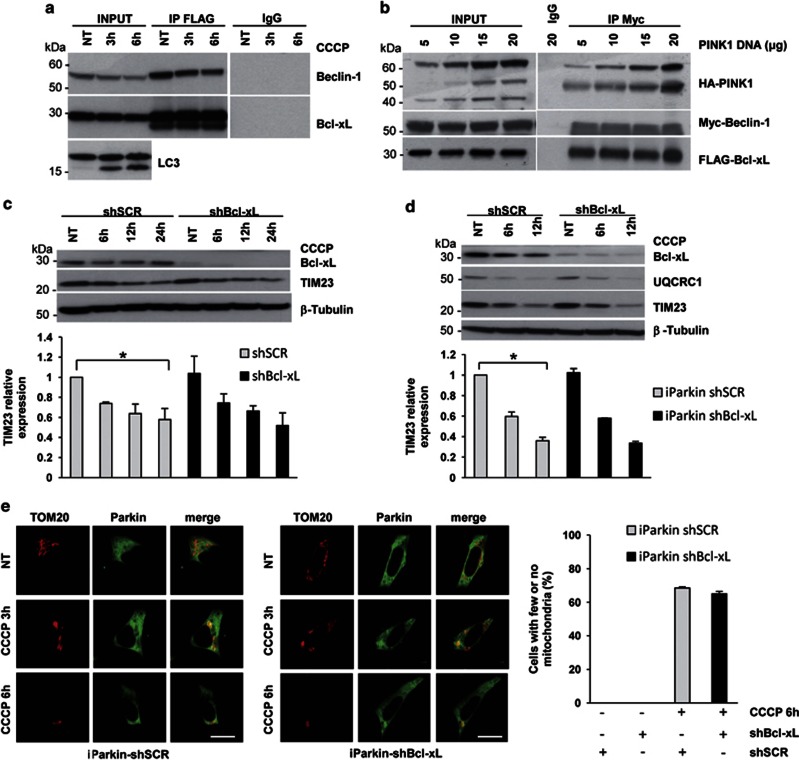
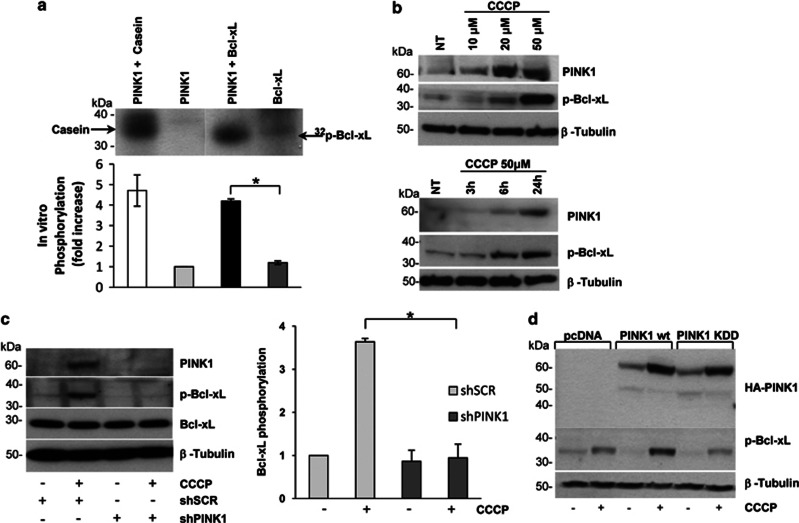
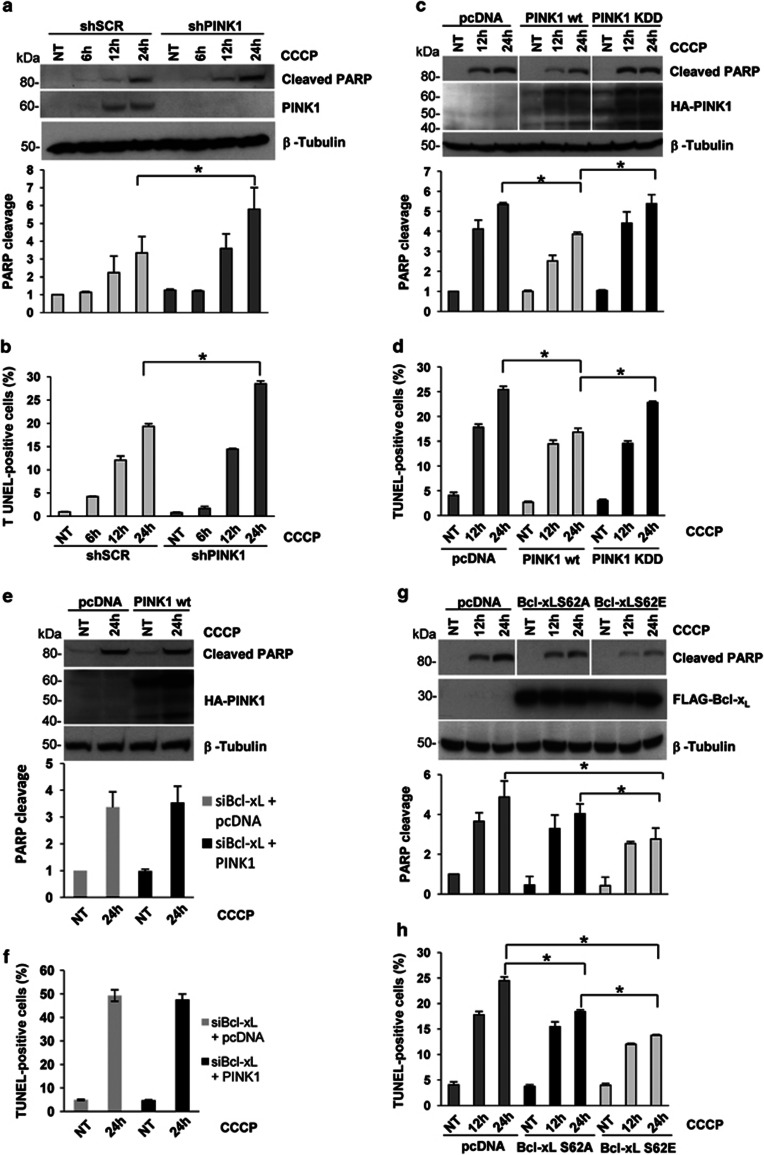
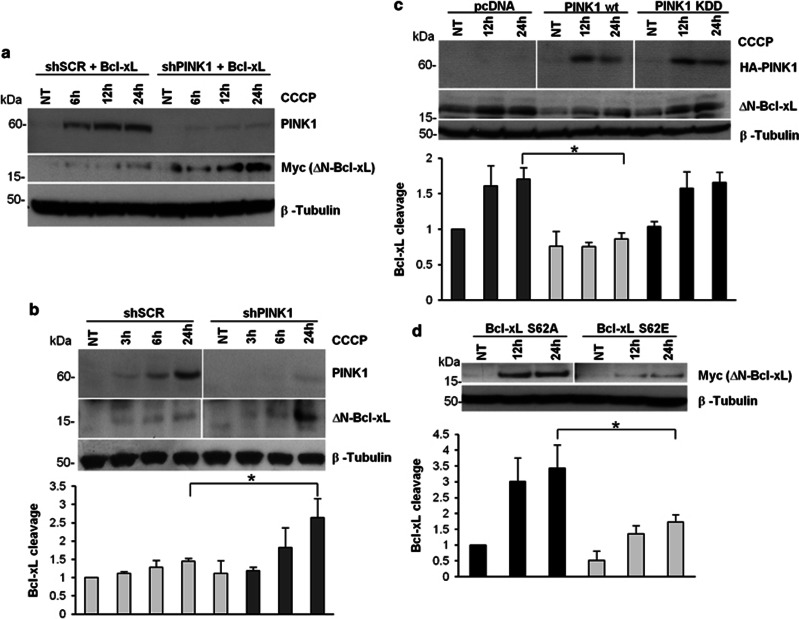
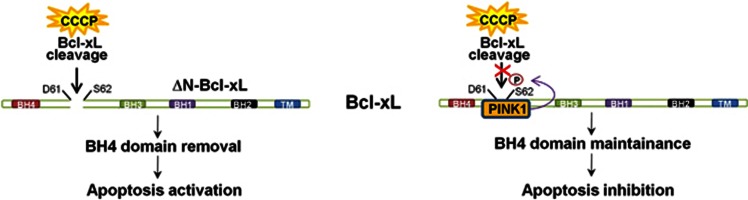
Mutations in the PINK1 gene are a frequent cause of autosomal recessive Parkinson's disease (PD). PINK1 encodes a mitochondrial kinase with neuroprotective activity, implicated in maintaining mitochondrial homeostasis and function. In concurrence with Parkin, PINK1 regulates mitochondrial trafficking and degradation of damaged mitochondria through mitophagy. Moreover, PINK1 can activate autophagy by interacting with the pro-autophagic protein Beclin-1. Here, we report that, upon mitochondrial depolarization, PINK1 interacts with and phosphorylates Bcl-xL, an anti-apoptotic protein also known to inhibit autophagy through its binding to Beclin-1. PINK1-Bcl-xL interaction does not interfere either with Beclin-1 release from Bcl-xL or the mitophagy pathway; rather it protects against cell death by hindering the pro-apoptotic cleavage of Bcl-xL. Our data provide a functional link between PINK1, Bcl-xL and apoptosis, suggesting a novel mechanism through which PINK1 regulates cell survival. This pathway could be relevant for the pathogenesis of PD as well as other diseases including cancer.
Figures
Similar articles
-
[Autophagy promoted by Pakinson's disease related protein Pink1].Sichuan Da Xue Xue Bao Yi Xue Ban. 2013 May;44(3):366-70. Sichuan Da Xue Xue Bao Yi Xue Ban. 2013. PMID: 23898514 Chinese.
-
Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy.Mol Cell. 2014 Aug 7;55(3):451-66. doi: 10.1016/j.molcel.2014.06.001. Epub 2014 Jul 3. Mol Cell. 2014. PMID: 24999239
-
Role of Bcl-xL/Beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death.Biochem Pharmacol. 2014 Mar 15;88(2):178-88. doi: 10.1016/j.bcp.2014.01.027. Epub 2014 Jan 31. Biochem Pharmacol. 2014. PMID: 24486574 Free PMC article.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
-
The mitochondrial kinase PINK1: functions beyond mitophagy.J Neurochem. 2016 Oct;139 Suppl 1:232-239. doi: 10.1111/jnc.13655. Epub 2016 Jun 2. J Neurochem. 2016. PMID: 27251035 Review.
Cited by
-
The Links between Parkinson's Disease and Cancer.Biomedicines. 2020 Oct 14;8(10):416. doi: 10.3390/biomedicines8100416. Biomedicines. 2020. PMID: 33066407 Free PMC article. Review.
-
Alpha-tocotrienol enhances arborization of primary hippocampal neurons via upregulation of Bcl-xL.Nutr Res. 2022 May;101:31-42. doi: 10.1016/j.nutres.2022.02.007. Epub 2022 Mar 7. Nutr Res. 2022. PMID: 35366596 Free PMC article.
-
Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice.Cell Death Discov. 2022 Jan 17;8(1):28. doi: 10.1038/s41420-022-00826-8. Cell Death Discov. 2022. PMID: 35039479 Free PMC article.
-
Mitochondria in health, disease, and aging.Physiol Rev. 2023 Oct 1;103(4):2349-2422. doi: 10.1152/physrev.00058.2021. Epub 2023 Apr 6. Physiol Rev. 2023. PMID: 37021870 Free PMC article. Review.
-
In Vitro Comparison of the Activity Requirements and Substrate Specificity of Human and Triboleum castaneum PINK1 Orthologues.PLoS One. 2016 Jan 19;11(1):e0146083. doi: 10.1371/journal.pone.0146083. eCollection 2016. PLoS One. 2016. PMID: 26784449 Free PMC article.
References
-
- Siderowf A, Stern M. Update on Parkinson disease. Ann Intern Med. 2003;138:651–658. - PubMed
-
- Schapira AH. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. - PubMed
-
- Belin AC, Westerlund M. Parkinson's disease: a genetic perspective. FEBS J. 2008;275:1377–1383. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
