

Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome
- PMID: 23522784
- PMCID: PMC3617382
- DOI: 10.1016/j.ajhg.2013.02.012
Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome
Abstract
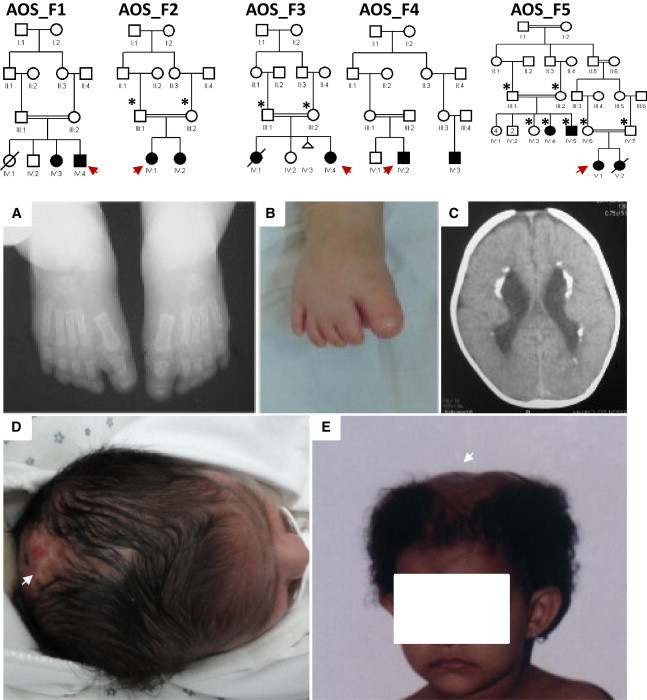
Adams-Oliver syndrome (AOS) is a rare, autosomal-dominant or -recessive disorder characterized primarily by aplasia cutis congenita and terminal transverse limb defects. Recently, we demonstrated that homozygous mutations in DOCK6 cause an autosomal-recessive form of AOS. In this study, we sought to determine the contribution of DOCK6 mutations to the etiology of AOS in several consanguineous families. In two of the five families studied, we identified two homozygous truncating mutations (a splice-site mutation and a frameshift duplication). DOCK6 sequencing revealed no mutation in the remaining three families, consistent with their autozygosity mapping and linkage-analysis results, which revealed a single candidate locus in 3p14.1 on three different haplotype backgrounds in the three families. Indeed, exome sequencing in one family revealed one missense mutation in EOGT (C3orf64), and subsequent targeted sequencing of this gene revealed a homozygous missense mutation and a homozygous frameshift deletion mutation in the other two families. EOGT encodes EGF-domain-specific O-linked N-acetylglucosamine (O-GlcNAc) transferase, which is involved in the O-GlcNAcylation (attachment of O-GlcNAc to serine and threonine residues) of a subset of extracellular EGF-domain-containing proteins. It has a documented role in epithelial-cell-matrix interactions in Drosophila, in which deficiency of its ortholog causes wing blistering. Our findings highlight a developmental role of O-GlcNAcylation in humans and expand the genetic heterogeneity of autosomal-recessive AOS.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Adams F.H., Oliver C.P. Hereditary deformities in man due to arrested development. J. Hered. 1945;36:3–7.
-
- Whitley C.B., Gorlin R.J. Adams-Oliver syndrome revisited. Am. J. Med. Genet. 1991;40:319–326. - PubMed
-
- Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. - PubMed
-
- Temtamy S.A., Aglan M.S., Ashour A.M., Zaki M.S. Adams-Oliver syndrome: further evidence of an autosomal recessive variant. Clin. Dysmorphol. 2007;16:141–149. - PubMed
-
- Temtamy S.A., McKusick V.A. Alan R. Liss; New York: 1978. The Genetics of Hand Malformations. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
