

Identification of cytokinin-responsive genes using microarray meta-analysis and RNA-Seq in Arabidopsis
- PMID: 23524861
- PMCID: PMC3641208
- DOI: 10.1104/pp.113.217026
Identification of cytokinin-responsive genes using microarray meta-analysis and RNA-Seq in Arabidopsis
Abstract
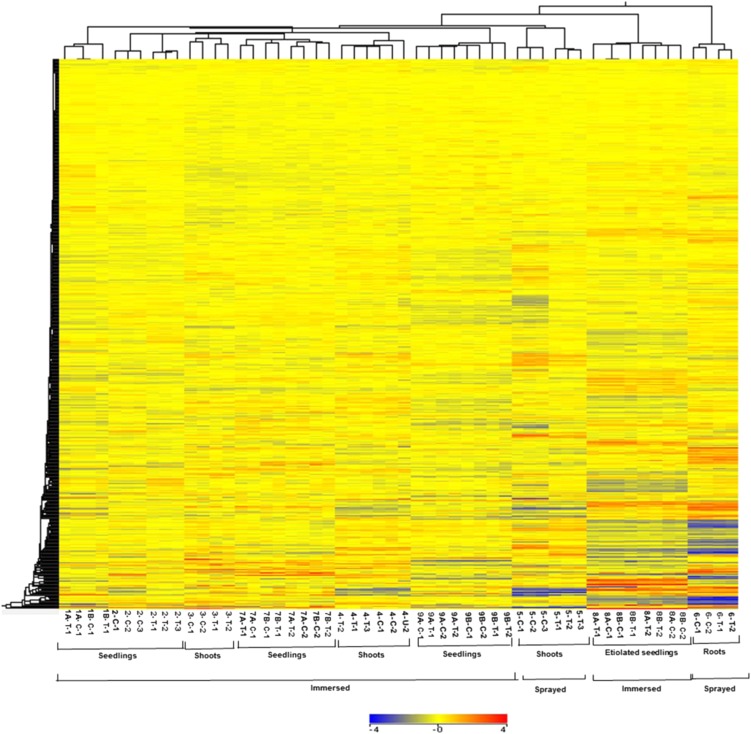
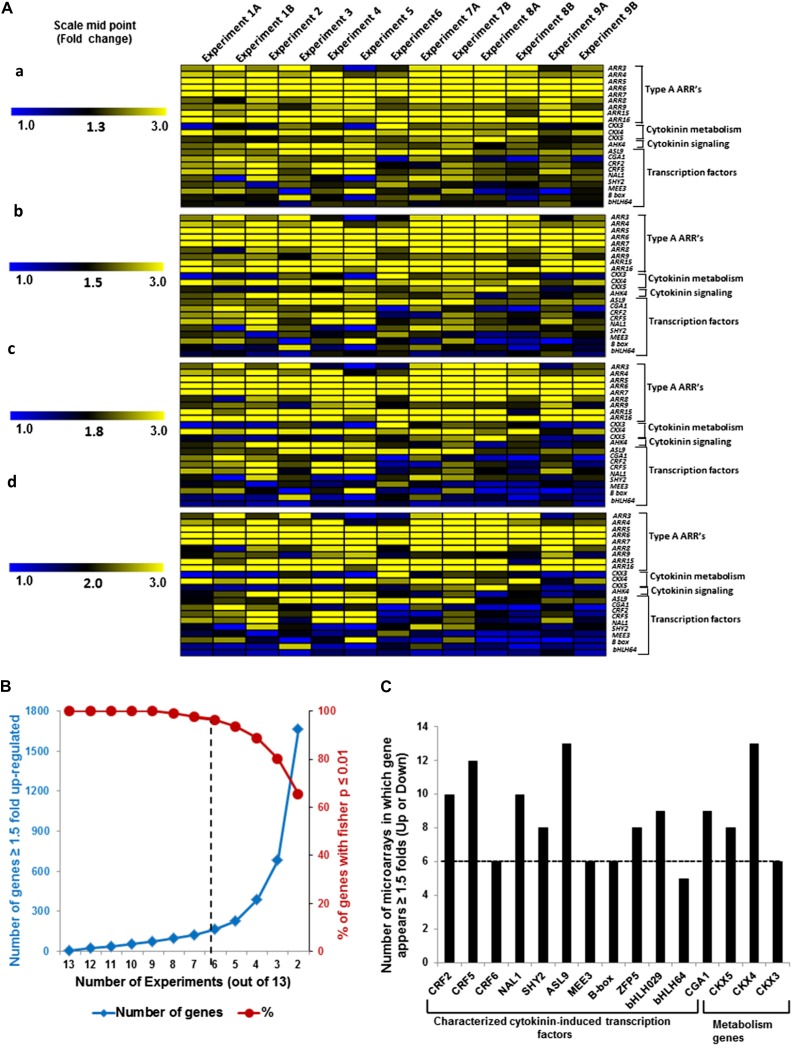
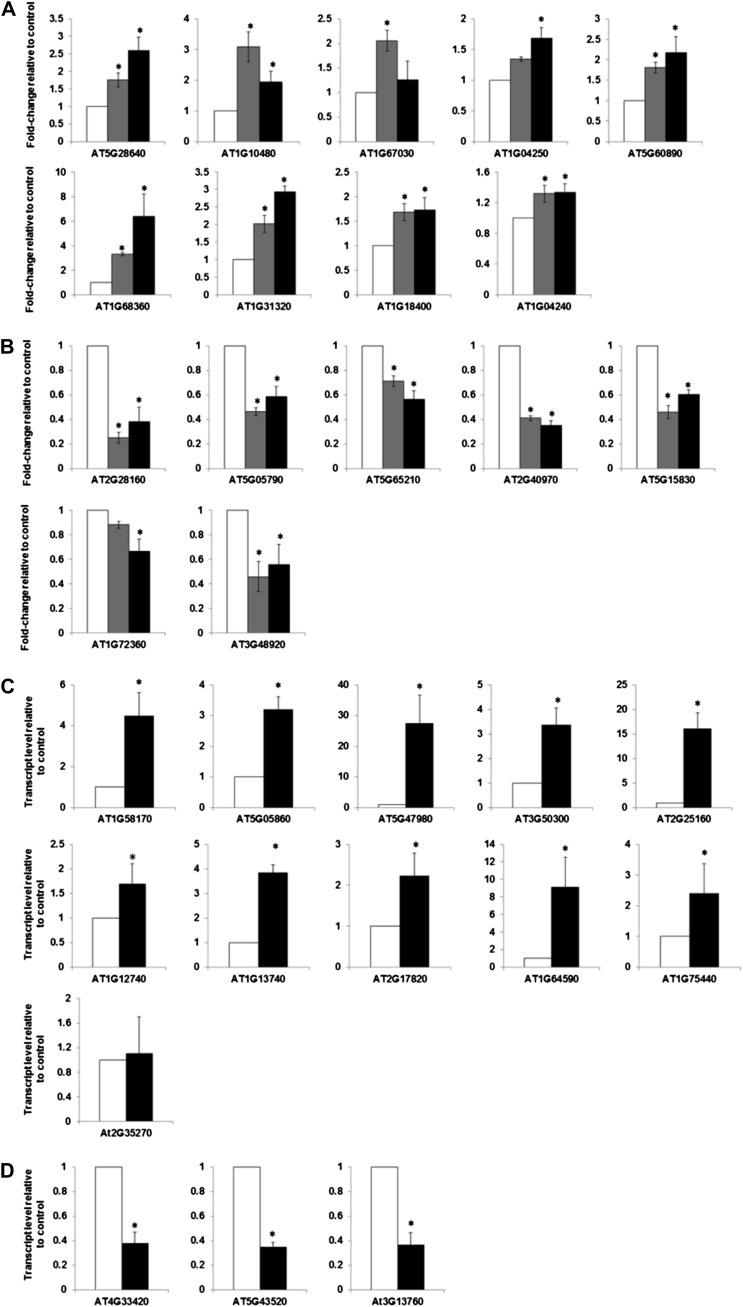
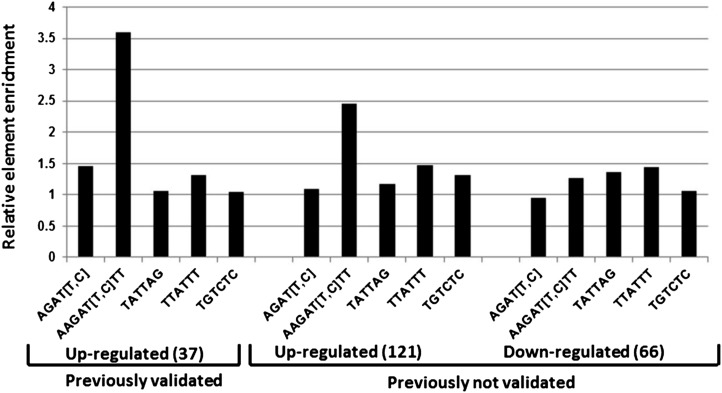
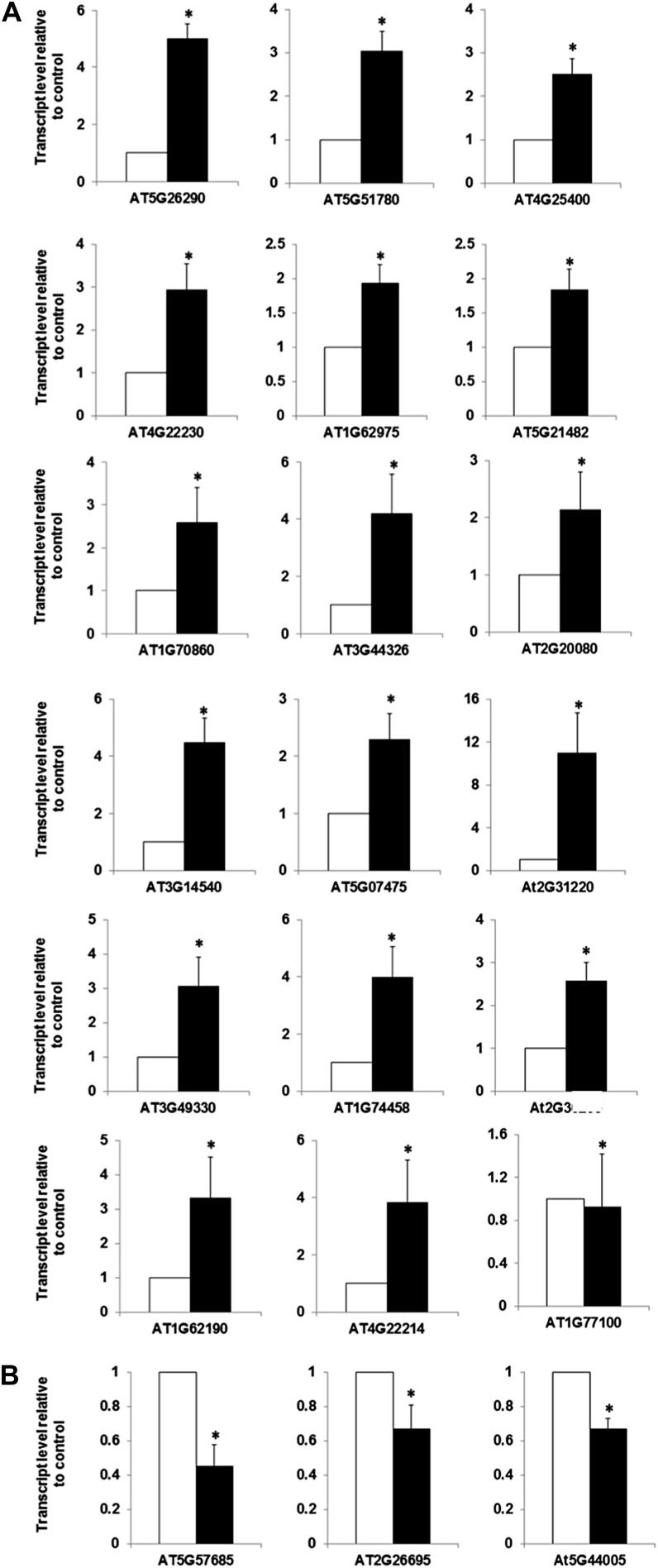
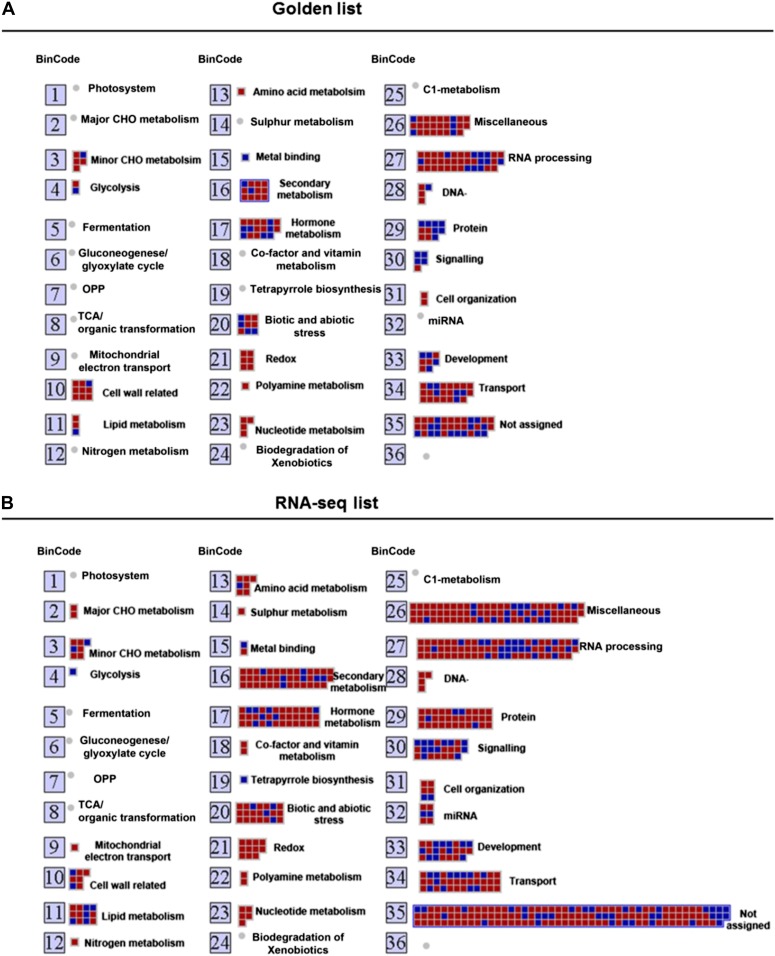
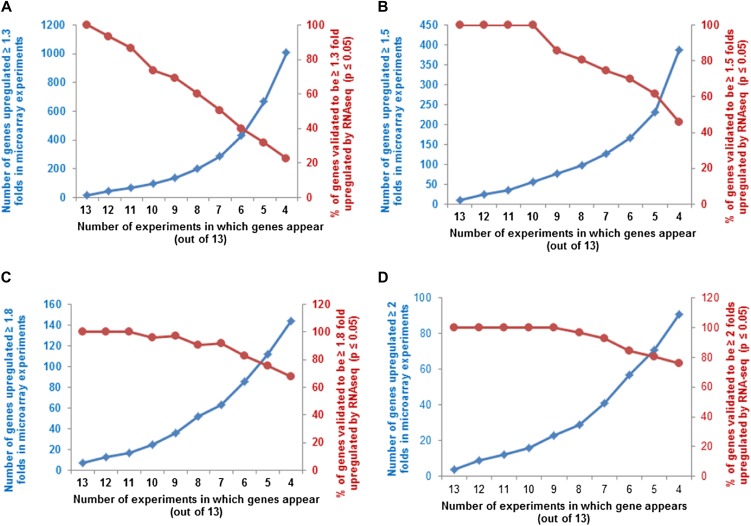
Cytokinins are N(6)-substituted adenine derivatives that play diverse roles in plant growth and development. We sought to define a robust set of genes regulated by cytokinin as well as to query the response of genes not represented on microarrays. To this end, we performed a meta-analysis of microarray data from a variety of cytokinin-treated samples and used RNA-seq to examine cytokinin-regulated gene expression in Arabidopsis (Arabidopsis thaliana). Microarray meta-analysis using 13 microarray experiments combined with empirically defined filtering criteria identified a set of 226 genes differentially regulated by cytokinin, a subset of which has previously been validated by other methods. RNA-seq validated about 73% of the up-regulated genes identified by this meta-analysis. In silico promoter analysis indicated an overrepresentation of type-B Arabidopsis response regulator binding elements, consistent with the role of type-B Arabidopsis response regulators as primary mediators of cytokinin-responsive gene expression. RNA-seq analysis identified 73 cytokinin-regulated genes that were not represented on the ATH1 microarray. Representative genes were verified using quantitative reverse transcription-polymerase chain reaction and NanoString analysis. Analysis of the genes identified reveals a substantial effect of cytokinin on genes encoding proteins involved in secondary metabolism, particularly those acting in flavonoid and phenylpropanoid biosynthesis, as well as in the regulation of redox state of the cell, particularly a set of glutaredoxin genes. Novel splicing events were found in members of some gene families that are known to play a role in cytokinin signaling or metabolism. The genes identified in this analysis represent a robust set of cytokinin-responsive genes that are useful in the analysis of cytokinin function in plants.
Figures

References
-
- An L, Zhou Z, Sun L, Yan A, Xi W, Yu N, Cai W, Chen X, Yu H, Schiefelbein J, et al. (2012) A zinc finger protein gene ZFP5 integrates phytohormone signaling to control root hair development in Arabidopsis. Plant J 72: 474–490 - PubMed
-
- Argueso CT, Ferreira FJ, Kieber JJ. (2009) Environmental perception avenues: the interaction of cytokinin and environmental response pathways. Plant Cell Environ 32: 1147–1160 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
