

Detailed molecular epidemiologic characterization of HIV-1 infection in Bulgaria reveals broad diversity and evolving phylodynamics
- PMID: 23527245
- PMCID: PMC3602066
- DOI: 10.1371/journal.pone.0059666
Detailed molecular epidemiologic characterization of HIV-1 infection in Bulgaria reveals broad diversity and evolving phylodynamics
Abstract
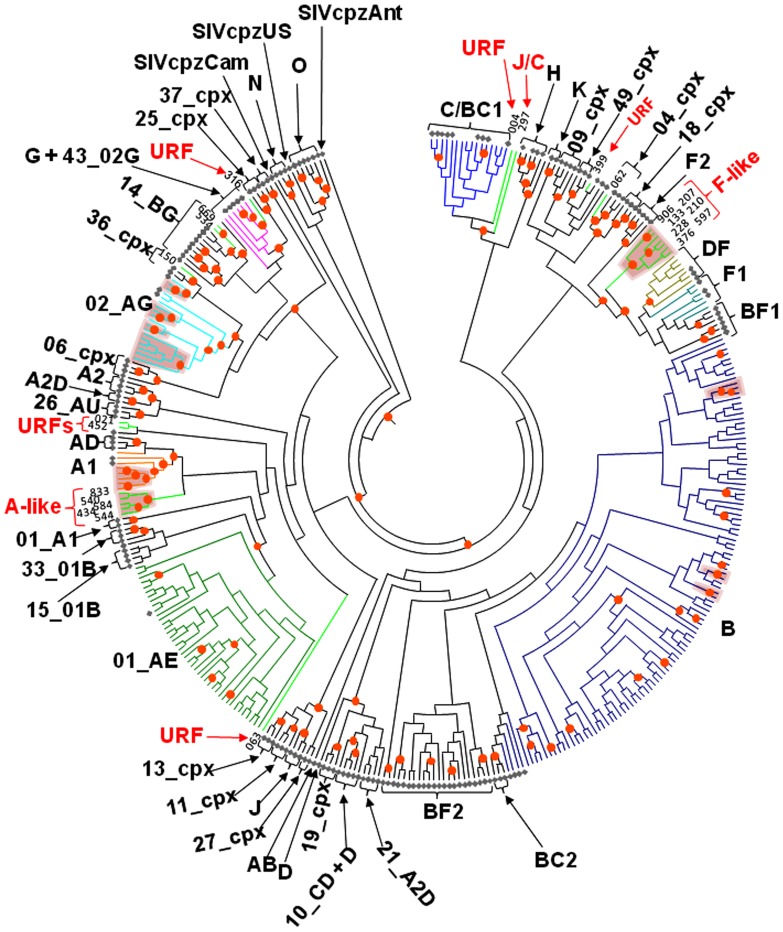
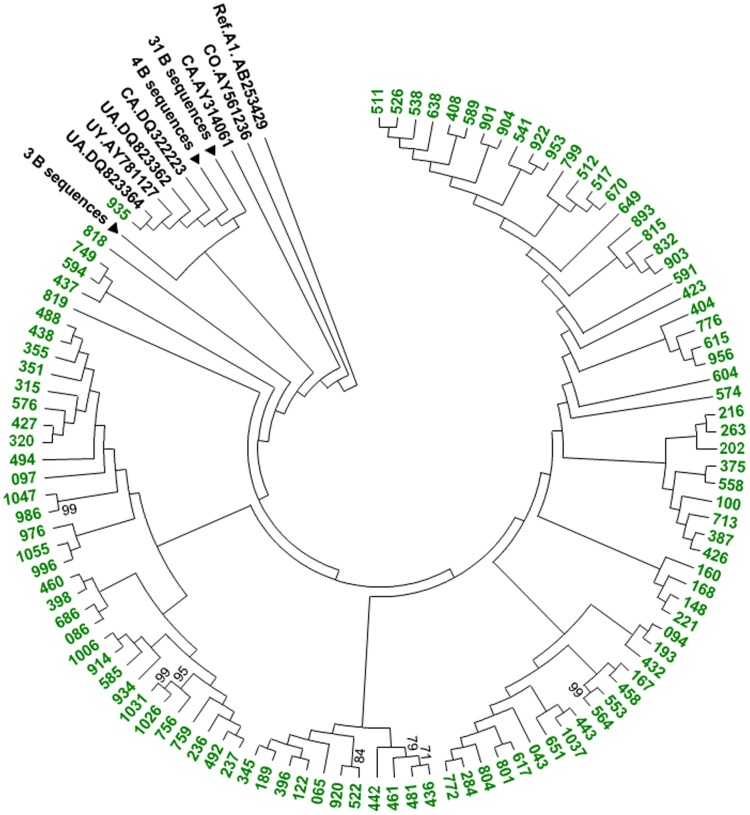
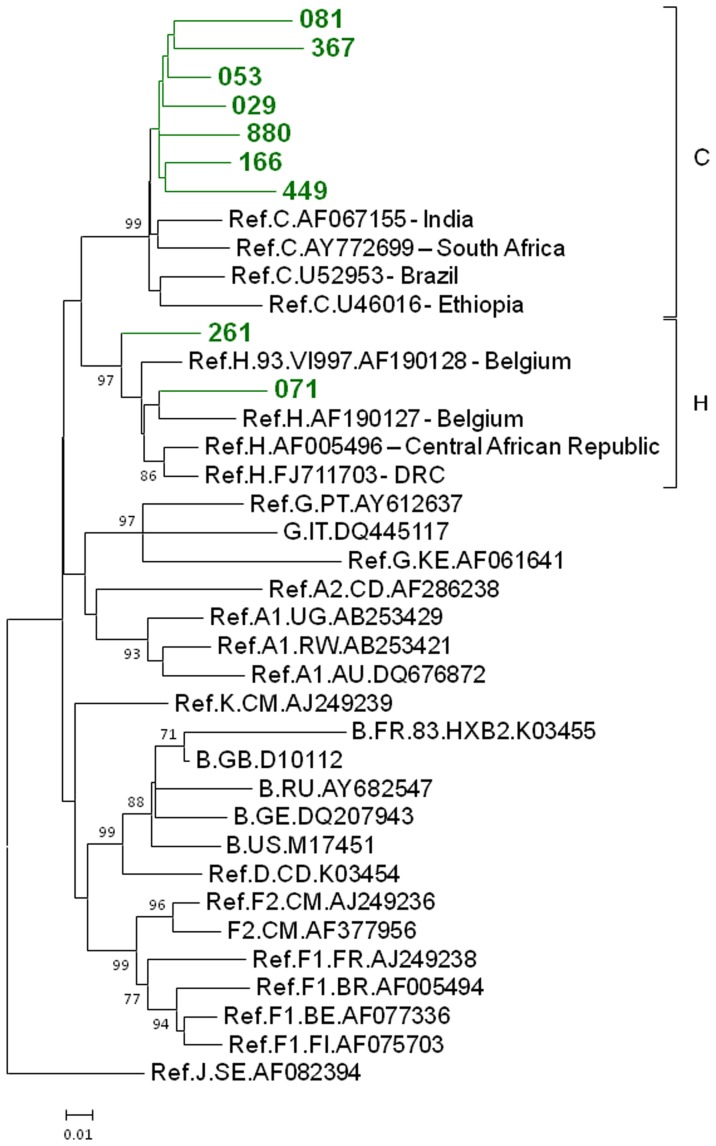
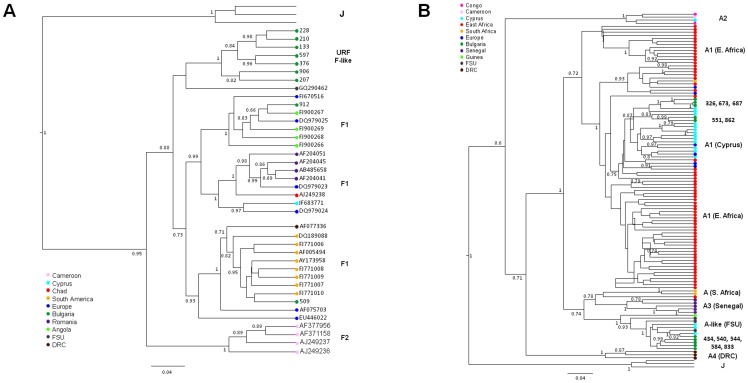
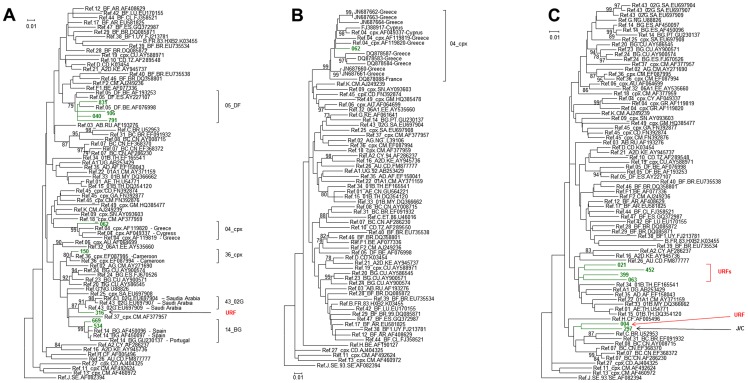
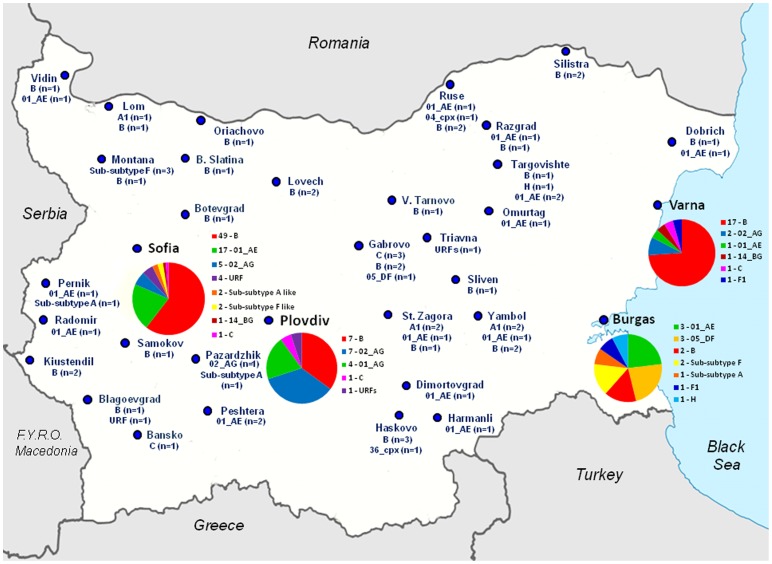
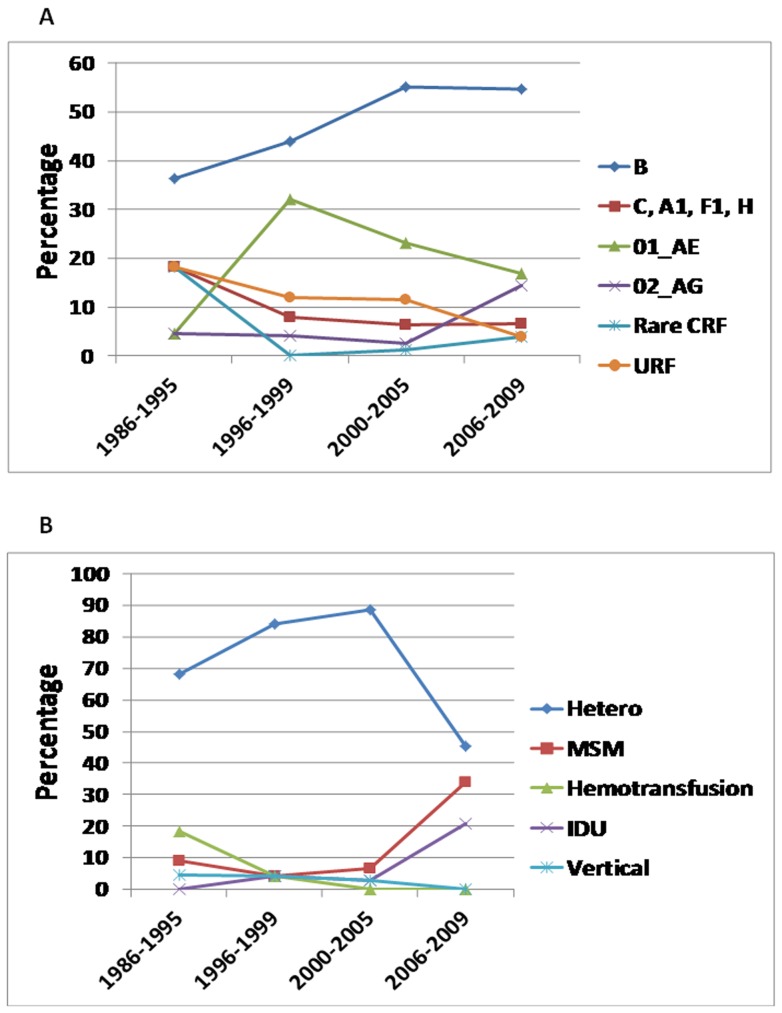
Limited information is available to describe the molecular epidemiology of HIV-1 in Bulgaria. To better understand the genetic diversity and the epidemiologic dynamics of HIV-1 we analyzed 125 new polymerase (pol) sequences from Bulgarians diagnosed through 2009 and 77 pol sequences available from our previous study from persons infected prior to 2007. Epidemiologic and demographic information was obtained from each participant and phylogenetic analysis was used to infer HIV-1 evolutionary histories. 120 (59.5%) persons were infected with one of five different HIV-1 subtypes (A1, B, C, F1 and H) and 63 (31.2%) persons were infected with one of six different circulating recombinant forms (CRFs; 01_AE, 02_AG, 04_cpx, 05_DF, 14_BG, and 36_cpx). We also for the first time identified infection with two different clusters of unique A-like and F-like sub-subtype variants in 12 persons (5.9%) and seven unique recombinant forms (3.5%), including a novel J/C recombinant. While subtype B was the major genotype identified and was more prevalent in MSM and increased between 2000-2005, most non-B subtypes were present in persons ≥45 years old. CRF01_AE was the most common non-B subtype and was higher in women and IDUs relative to other risk groups combined. Our results show that HIV-1 infection in Bulgaria reflects the shifting distribution of genotypes coincident with the changing epidemiology of the HIV-1 epidemic among different risk groups. Our data support increased public health interventions targeting IDUs and MSM. Furthermore, the substantial and increasing HIV-1 genetic heterogeneity, combined with fluctuating infection dynamics, highlights the importance of sustained and expanded surveillance to prevent and control HIV-1 infection in Bulgaria.
Conflict of interest statement
Figures

References
-
- Plantier JC, Leoz M, Dickerson JE, De Oliveira F, Cordonnier F, et al. (2009) A new human immunodeficiency virus derived from gorillas. Nat Med 15: 871–872. - PubMed
-
- Hemelaar J (2012) The origin and diversity of the HIV-1 pandemic. Trends in Molecular Medicine 18: 182–192. - PubMed
-
- Louwagie J, McCutchan FE, Peeters M, Brennan TP, Sanders-Buell E, et al. (1993) Phylogenetic analysis of gag genes from 70 international HIV-1 isolates provides evidence for multiple genotypes. AIDS 7: 769–780. - PubMed
-
- Olsen GJ, Matsuda H, Hagstrom R, Overbeek R (2012) fastDNAml: A tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci 10: 41–8. - PubMed
-
- Alaeus A (2000) Significance of HIV-1 genetic subtypes. Scand J Infect Dis 32: 455–463. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
