

Cathepsin K knockout alleviates pressure overload-induced cardiac hypertrophy
- PMID: 23529168
- PMCID: PMC3929275
- DOI: 10.1161/HYPERTENSIONAHA.111.00947
Cathepsin K knockout alleviates pressure overload-induced cardiac hypertrophy
Abstract
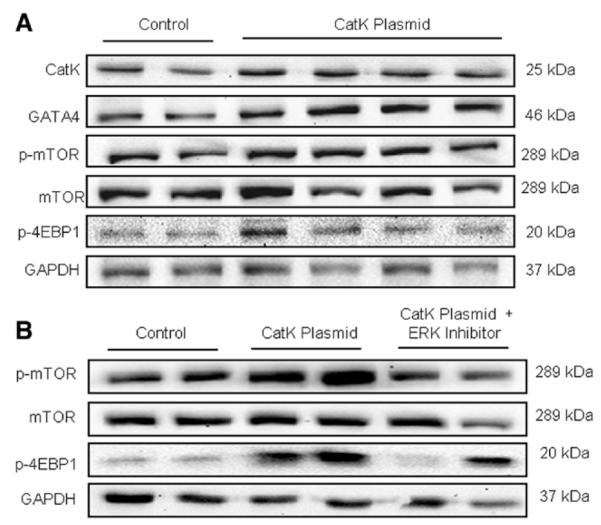
Evidence from human and animal studies has documented elevated levels of lysosomal cysteine protease cathepsin K in failing hearts. Here, we hypothesized that ablation of cathepsin K mitigates pressure overload-induced cardiac hypertrophy. Cathepsin K knockout mice and their wild-type littermates were subjected to abdominal aortic constriction, resulting in cardiac remodeling (heart weight, cardiomyocyte size, left ventricular wall thickness, and end diastolic and end systolic dimensions) and decreased fractional shortening, the effects of which were significantly attenuated or ablated by cathepsin K knockout. Pressure overload dampened cardiomyocyte contractile function along with decreased resting Ca2+ levels and delayed Ca2+ clearance, which were partly resolved by cathepsin K knockout. Cardiac mammalian target of rapamycin and extracellular signal-regulated kinases (ERK) signaling cascades were upregulated by pressure overload, the effects of which were attenuated by cathepsin K knockout. In cultured H9c2 myoblast cells, silencing of cathepsin K blunted, whereas cathepsin K transfection mimicked phenylephrine-induced hypertrophic response, along with elevated phosphorylation of mammalian target of rapamycin and ERK. In addition, cathepsin K protein levels were markedly elevated in human hearts of end-stage dilated cardiomyopathy. Collectively, our data suggest that cathepsin K ablation mitigates pressure overload-induced hypertrophy, possibly via inhibition of the mammalian target of rapamycin and ERK pathways.
Keywords: cardiac hypertrophy; cathepsin K; contractile function; mammalian target of rapamycin.
Figures









Similar articles
-
Cathepsin K knockout alleviates aging-induced cardiac dysfunction.Aging Cell. 2015 Jun;14(3):345-51. doi: 10.1111/acel.12276. Epub 2015 Feb 18. Aging Cell. 2015. PMID: 25692548 Free PMC article.
-
Cathepsin K knockout mitigates high-fat diet-induced cardiac hypertrophy and contractile dysfunction.Diabetes. 2013 Feb;62(2):498-509. doi: 10.2337/db12-0350. Epub 2012 Oct 15. Diabetes. 2013. PMID: 23069627 Free PMC article.
-
CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload.Int J Cardiol. 2013 Oct 3;168(3):2049-56. doi: 10.1016/j.ijcard.2013.01.165. Epub 2013 Feb 26. Int J Cardiol. 2013. PMID: 23452880
-
Integration of pathways that signal cardiac growth with modulation of myofilament activity.J Nucl Cardiol. 2002 Sep-Oct;9(5):523-33. doi: 10.1067/mnc.2002.127626. J Nucl Cardiol. 2002. PMID: 12360133 Review.
-
Critical roles of macrophages in pressure overload-induced cardiac remodeling.J Mol Med (Berl). 2021 Jan;99(1):33-46. doi: 10.1007/s00109-020-02002-w. Epub 2020 Oct 31. J Mol Med (Berl). 2021. PMID: 33130927 Review.
Cited by
-
Cathepsin K knockout protects against cardiac dysfunction in diabetic mice.Sci Rep. 2017 Aug 18;7(1):8703. doi: 10.1038/s41598-017-09037-z. Sci Rep. 2017. PMID: 28821796 Free PMC article.
-
Melatonin alleviates angiotensin-II-induced cardiac hypertrophy via activating MICU1 pathway.Aging (Albany NY). 2020 Nov 26;13(1):493-515. doi: 10.18632/aging.202159. Epub 2020 Nov 26. Aging (Albany NY). 2020. PMID: 33259334 Free PMC article.
-
Cathepsin K as a key regulator of myocardial fibrosis in dilated cardiomyopathy and a promising therapeutic target.J Biol Chem. 2025 Aug;301(8):110421. doi: 10.1016/j.jbc.2025.110421. Epub 2025 Jun 25. J Biol Chem. 2025. PMID: 40578558 Free PMC article.
-
Multiple sites on SARS-CoV-2 spike protein are susceptible to proteolysis by cathepsins B, K, L, S, and V.Protein Sci. 2021 Jun;30(6):1131-1143. doi: 10.1002/pro.4073. Epub 2021 Apr 15. Protein Sci. 2021. PMID: 33786919 Free PMC article.
-
Cathepsin K knockout alleviates aging-induced cardiac dysfunction.Aging Cell. 2015 Jun;14(3):345-51. doi: 10.1111/acel.12276. Epub 2015 Feb 18. Aging Cell. 2015. PMID: 25692548 Free PMC article.
References
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. - PubMed
-
- Sam F, Siwik DA. Digesting the remodeled heart: role of lysosomal cysteine proteases in heart failure. Hypertension. 2006;48:830–831. - PubMed
-
- Cheng XW, Obata K, Kuzuya M, et al. Elastolytic cathepsin induction/activation system exists in myocardium and is upregulated in hypertensive heart failure. Hypertension. 2006;48:979–987. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
