

Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection
- PMID: 23536676
- PMCID: PMC3648129
- DOI: 10.1128/JVI.03557-12
Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection
Abstract
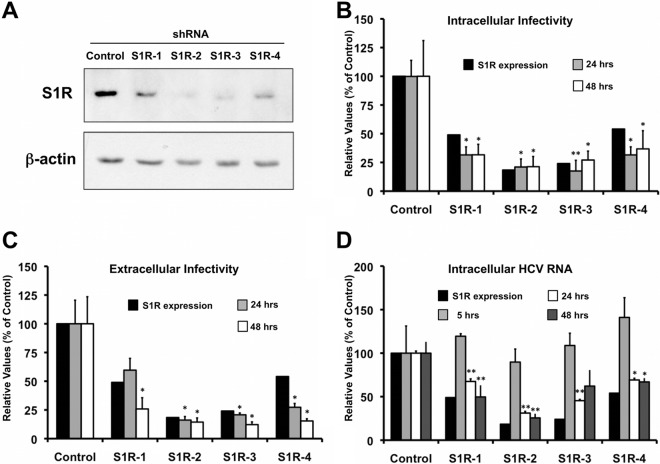
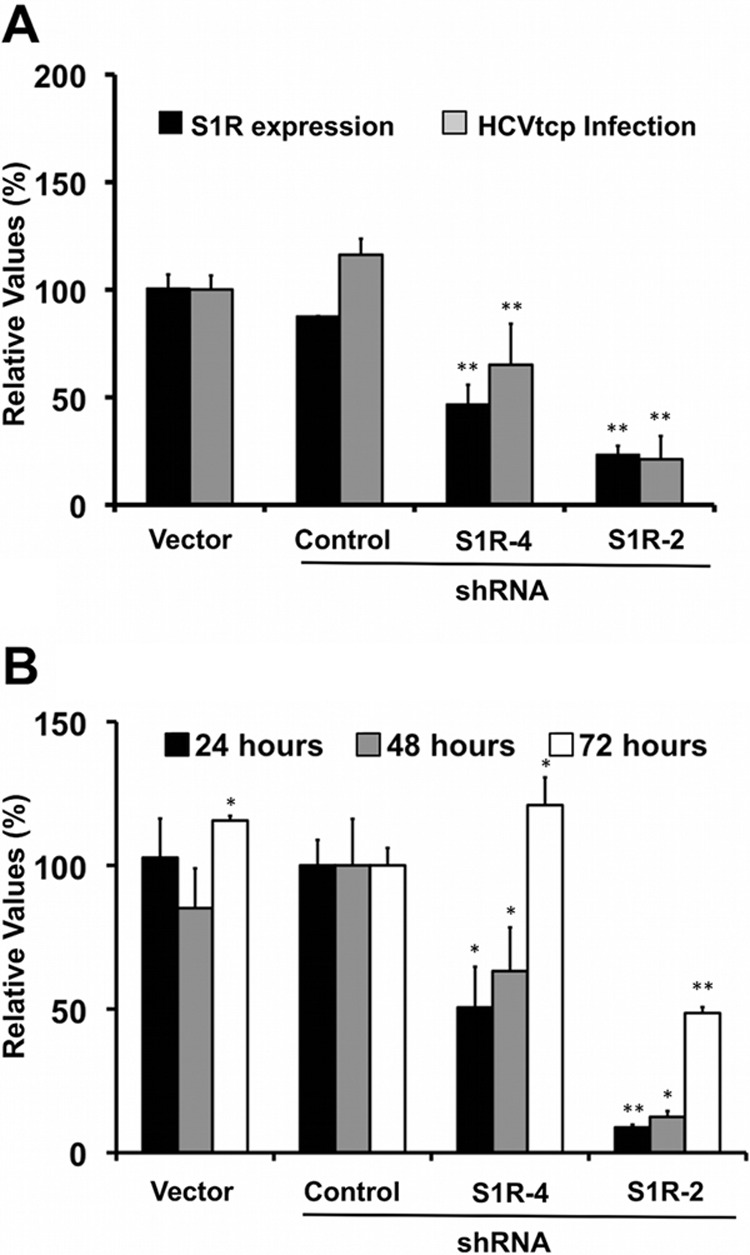
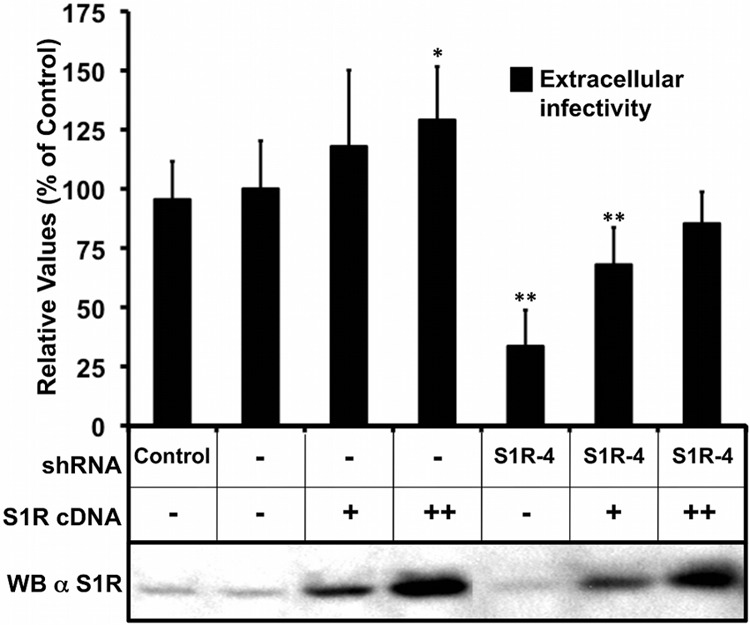
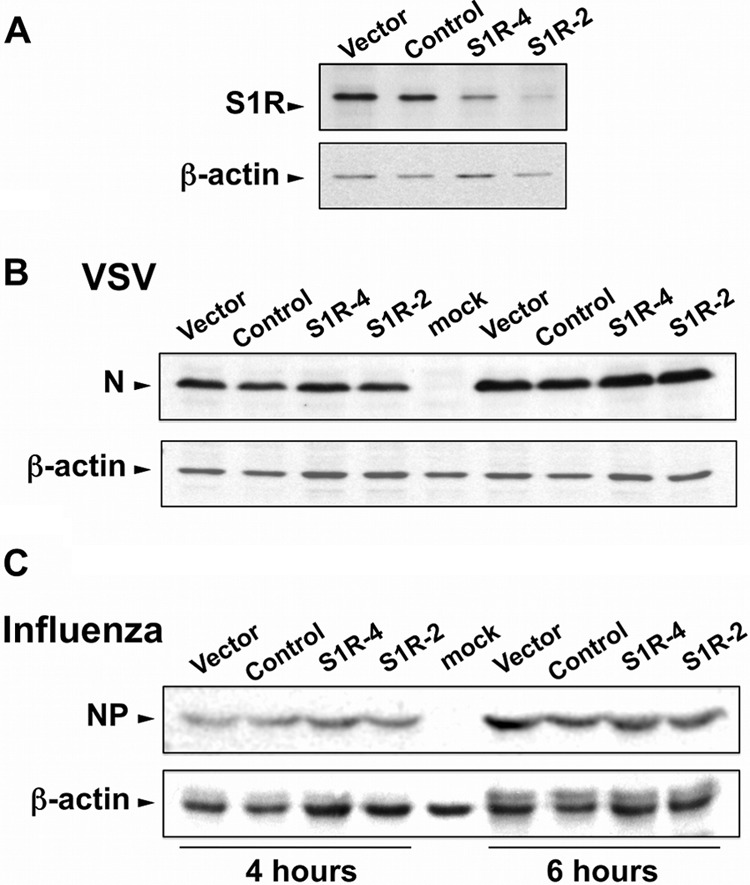
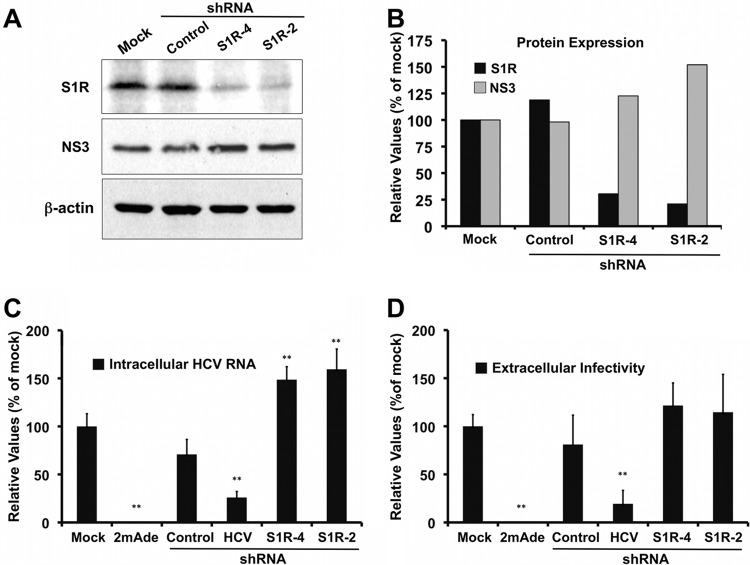
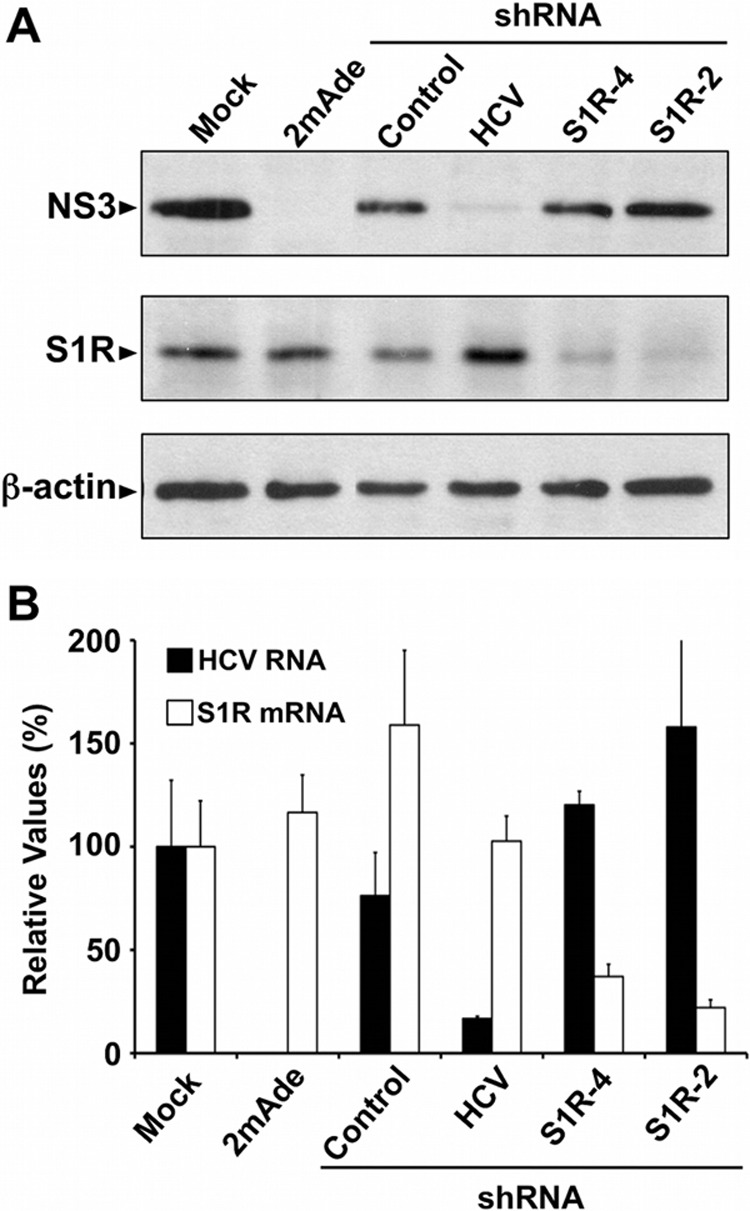
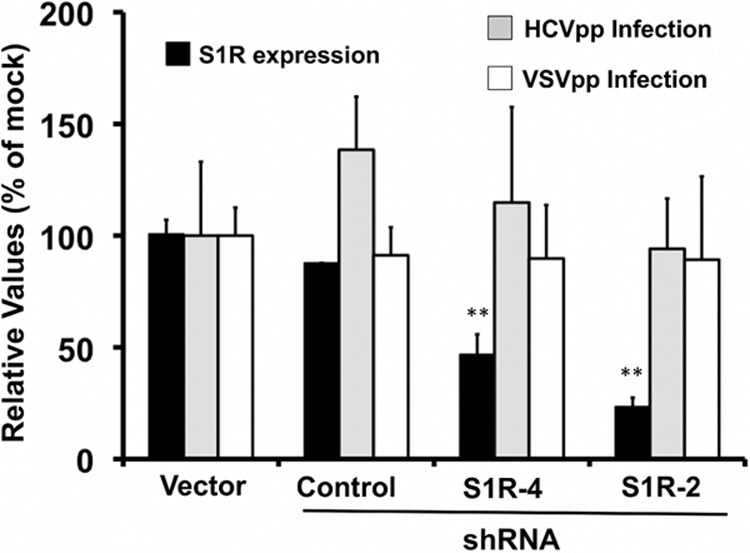
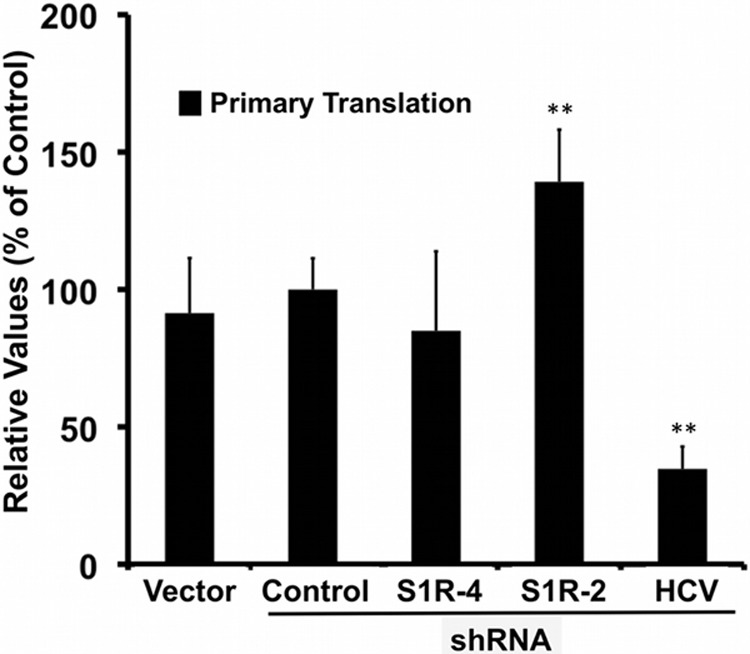
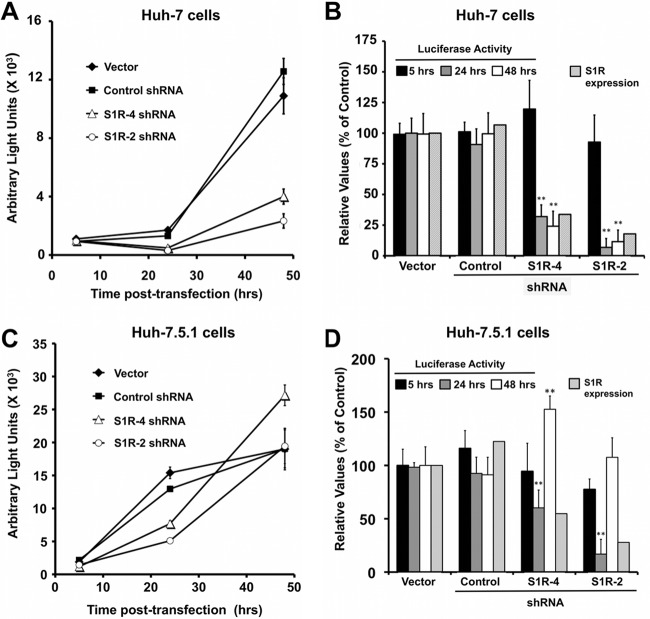
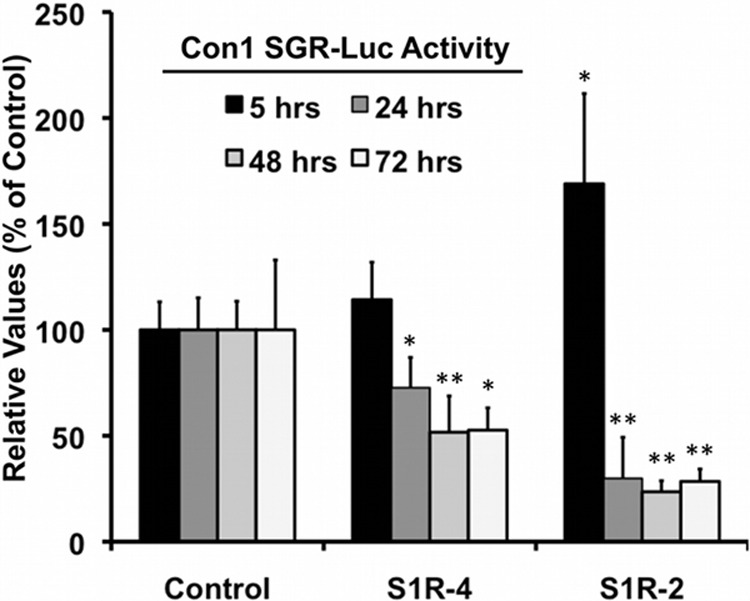
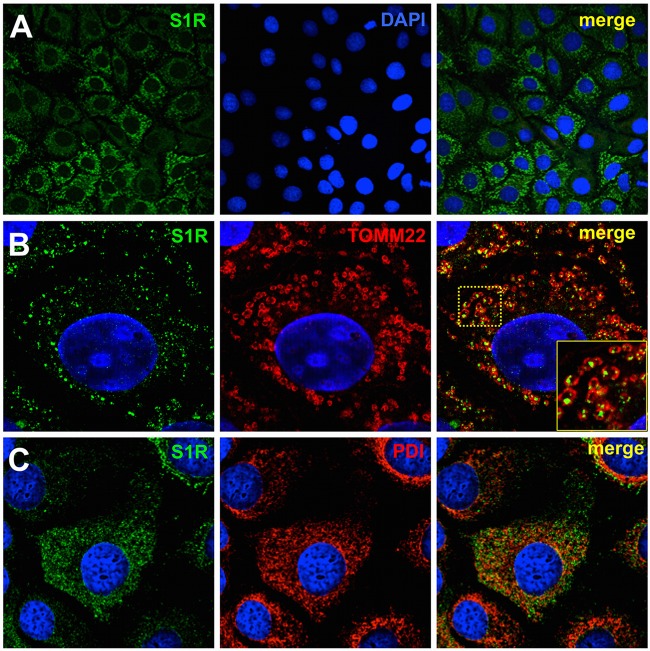
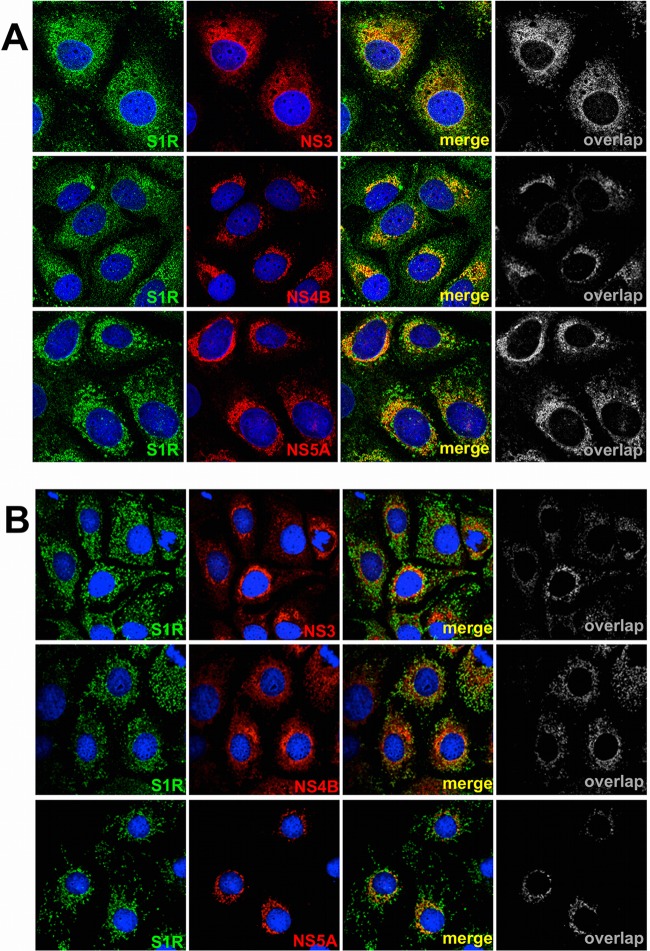
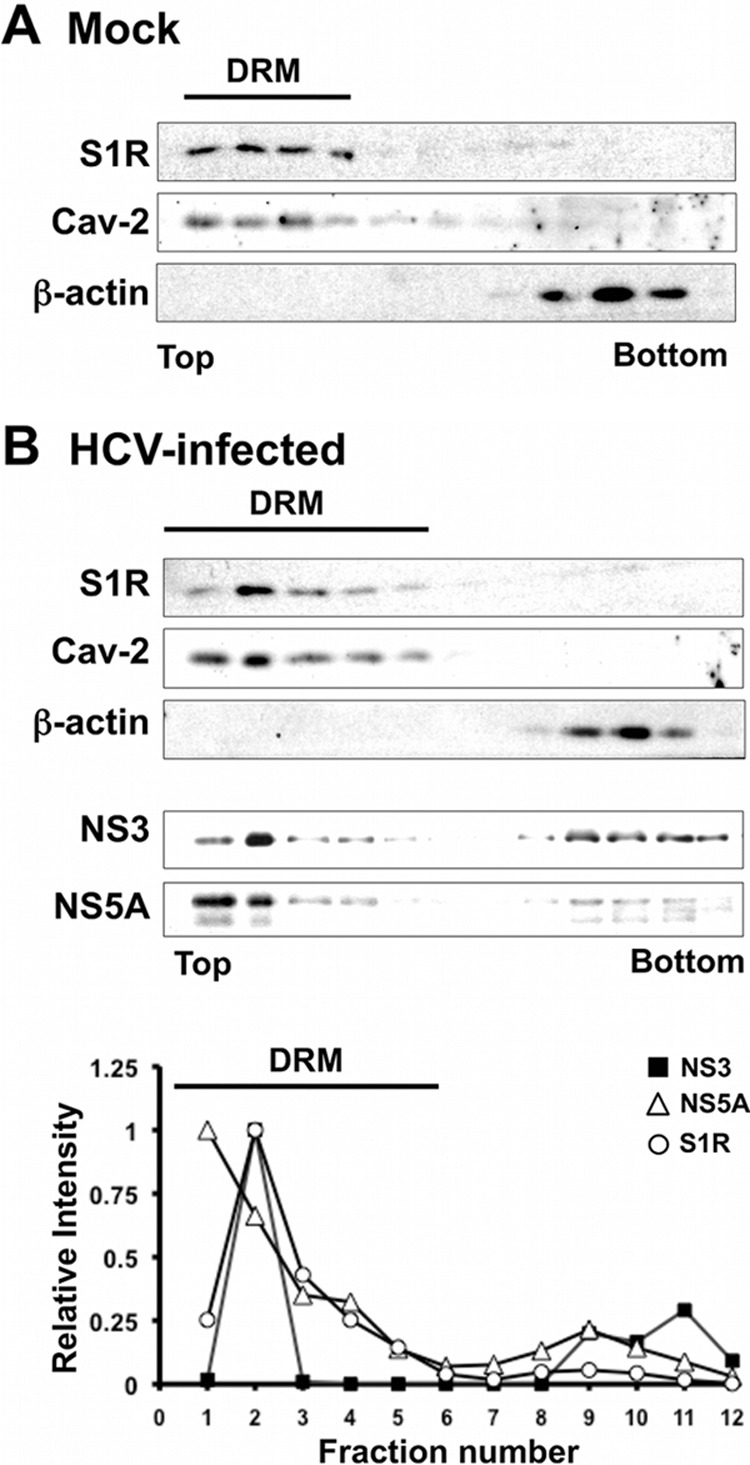
Hepatitis C virus (HCV) genome replication is thought to occur in a membranous cellular compartment derived from the endoplasmic reticulum (ER). The molecular mechanisms by which these membrane-associated replication complexes are formed during HCV infection are only starting to be unraveled, and both viral and cellular factors contribute to their formation. In this study, we describe the discovery of nonopioid sigma-1 receptor (S1R) as a cellular factor that mediates the early steps of viral RNA replication. S1R is a cholesterol-binding protein that resides in lipid-rich areas of the ER and in mitochondrion-associated ER membranes (MAMs). Several functions have been ascribed to this ER-resident chaperone, many of which are related to Ca(2+) signaling at the MAMs and lipid storage and trafficking. Downregulation of S1R expression by RNA interference (RNAi) in Huh-7 cells leads to a proportional decrease in susceptibility to HCV infection, as shown by reduced HCV RNA accumulation and intra- and extracellular infectivity in single-cycle infection experiments. Similar RNAi studies in persistently infected cells indicate that S1R expression is not rate limiting for persistent HCV RNA replication, as marked reduction in S1R in these cells does not lead to any decrease in HCV RNA or viral protein expression. However, subgenomic replicon transfection experiments indicate that S1R expression is rate limiting for HCV RNA replication without impairing primary translation. Overall, our data indicate that the initial steps of HCV infection are regulated by S1R, a key component of MAMs, suggesting that these structures could serve as platforms for initial RNA replication during HCV infection.
Figures
References
-
- Guidotti LG, Chisari FV. 2006. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1:23–61 - PubMed
-
- Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 - PubMed
-
- Otto GA, Puglisi JD. 2004. The pathway of HCV IRES-mediated translation initiation. Cell 119:369–380 - PubMed
-
- Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
