

Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures
- PMID: 23536761
- PMCID: PMC3594228
- DOI: 10.1371/journal.pone.0057148
Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures
Erratum in
- PLoS One. 2013;8(8). doi:10.1371/annotation/48a011e6-e4d0-4706-9a28-857eba8cfb31. Berry, Gerard [corrected to Berry, Gerard T]
Abstract
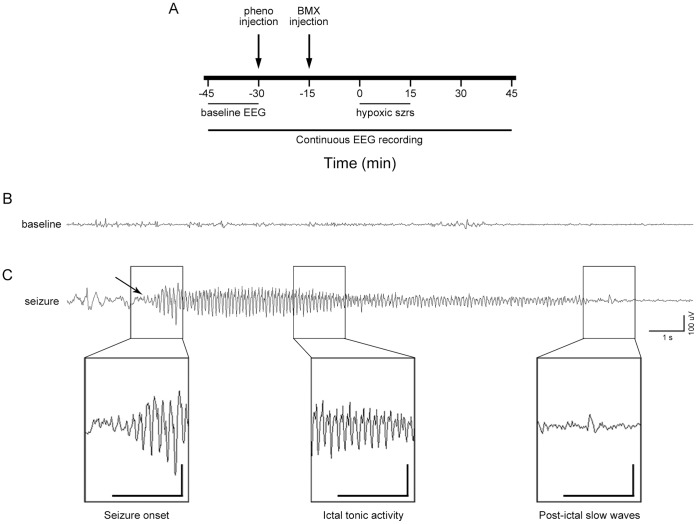
Neonatal seizures can be refractory to conventional anticonvulsants, and this may in part be due to a developmental increase in expression of the neuronal Na(+)-K(+)-2 Cl(-) cotransporter, NKCC1, and consequent paradoxical excitatory actions of GABAA receptors in the perinatal period. The most common cause of neonatal seizures is hypoxic encephalopathy, and here we show in an established model of neonatal hypoxia-induced seizures that the NKCC1 inhibitor, bumetanide, in combination with phenobarbital is significantly more effective than phenobarbital alone. A sensitive mass spectrometry assay revealed that bumetanide concentrations in serum and brain were dose-dependent, and the expression of NKCC1 protein transiently increased in cortex and hippocampus after hypoxic seizures. Importantly, the low doses of phenobarbital and bumetanide used in the study did not increase constitutive apoptosis, alone or in combination. Perforated patch clamp recordings from ex vivo hippocampal slices removed following seizures revealed that phenobarbital and bumetanide largely reversed seizure-induced changes in EGABA. Taken together, these data provide preclinical support for clinical trials of bumetanide in human neonates at risk for hypoxic encephalopathy and seizures.
Conflict of interest statement
Figures






References
-
- Ronen GM, Buckley D, Penney S, Streiner DL (2007) Long-term prognosis in children with neonatal seizures: a population-based study. Neurology 69: 1816–1822. - PubMed
-
- Sankar R, Painter MJ (2005) Neonatal seizures: After all these years we still love what doesn’t work. Neurology 64: 776–777. - PubMed
-
- Scher MS, Alvin J, Gaus L, Minnigh B, Painter MJ (2003) Uncoupling of EEG-clinical neonatal seizures after antiepileptic drug use. PediatrNeurol 28: 277–280. - PubMed
-
- Silverstein FS, Jensen FE (2007) Neonatal seizures. AnnNeurol 62: 112–120. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
