

Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing
- PMID: 23537139
- PMCID: PMC3706896
- DOI: 10.1186/gm432
Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing
Abstract
Background: To facilitate the clinical implementation of genomic medicine by next-generation sequencing, it will be critically important to obtain accurate and consistent variant calls on personal genomes. Multiple software tools for variant calling are available, but it is unclear how comparable these tools are or what their relative merits in real-world scenarios might be.
Methods: We sequenced 15 exomes from four families using commercial kits (Illumina HiSeq 2000 platform and Agilent SureSelect version 2 capture kit), with approximately 120X mean coverage. We analyzed the raw data using near-default parameters with five different alignment and variant-calling pipelines (SOAP, BWA-GATK, BWA-SNVer, GNUMAP, and BWA-SAMtools). We additionally sequenced a single whole genome using the sequencing and analysis pipeline from Complete Genomics (CG), with 95% of the exome region being covered by 20 or more reads per base. Finally, we validated 919 single-nucleotide variations (SNVs) and 841 insertions and deletions (indels), including similar fractions of GATK-only, SOAP-only, and shared calls, on the MiSeq platform by amplicon sequencing with approximately 5000X mean coverage.
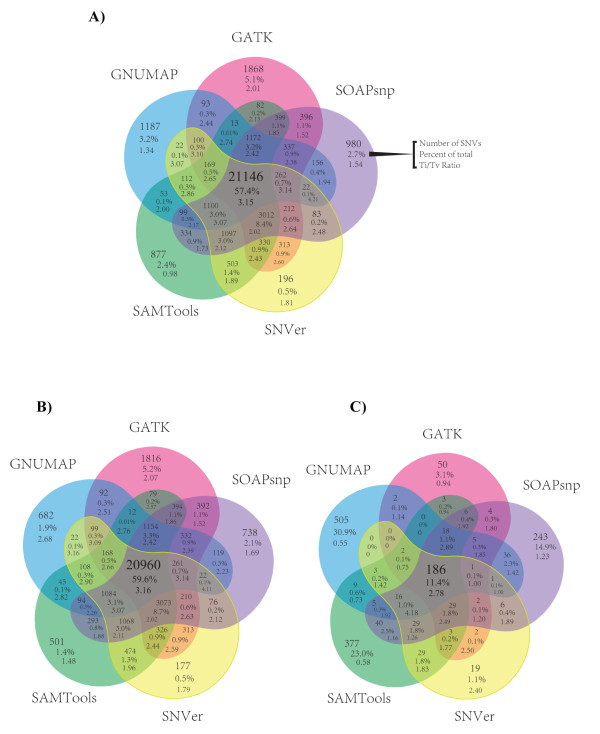
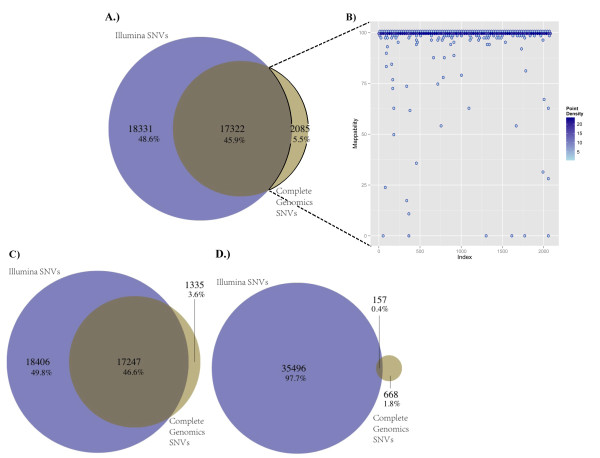
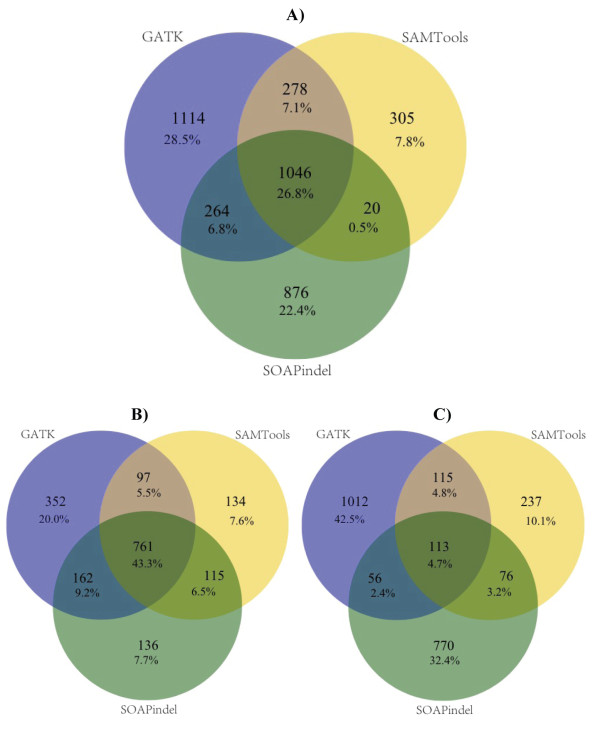
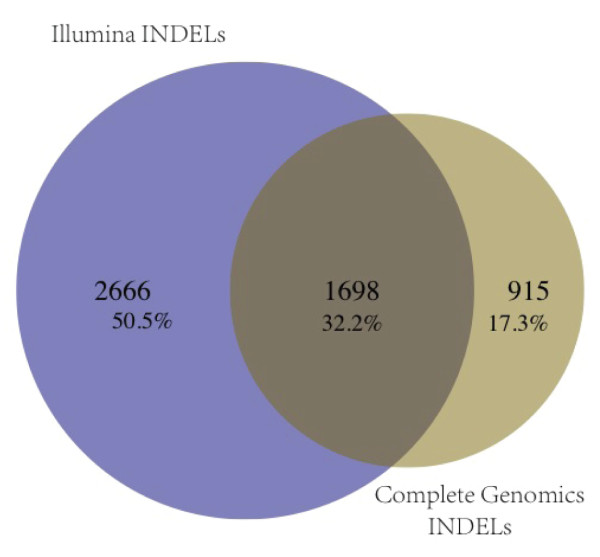
Results: SNV concordance between five Illumina pipelines across all 15 exomes was 57.4%, while 0.5 to 5.1% of variants were called as unique to each pipeline. Indel concordance was only 26.8% between three indel-calling pipelines, even after left-normalizing and intervalizing genomic coordinates by 20 base pairs. There were 11% of CG variants falling within targeted regions in exome sequencing that were not called by any of the Illumina-based exome analysis pipelines. Based on targeted amplicon sequencing on the MiSeq platform, 97.1%, 60.2%, and 99.1% of the GATK-only, SOAP-only and shared SNVs could be validated, but only 54.0%, 44.6%, and 78.1% of the GATK-only, SOAP-only and shared indels could be validated. Additionally, our analysis of two families (one with four individuals and the other with seven), demonstrated additional accuracy gained in variant discovery by having access to genetic data from a multi-generational family.
Conclusions: Our results suggest that more caution should be exercised in genomic medicine settings when analyzing individual genomes, including interpreting positive and negative findings with scrutiny, especially for indels. We advocate for renewed collection and sequencing of multi-generational families to increase the overall accuracy of whole genomes.
Figures

References
-
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science. 2012. - PMC - PubMed
-
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zollner S, Whittaker JC, Chissoe SL, Novembre J. et al.An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. - DOI - PMC - PubMed
-
- Bearn AG. Archibald Garrod and the individuality of Man. Oxford, New York: Clarendon Press; Oxford University Press; 1993.
-
- Ball MP, Thakuria JV, Zaranek AW, Clegg T, Rosenbaum AM, Wu X, Angrist M, Bhak J, Bobe J, Callow MJ, Cano C, Chou MF, Chung WK, Douglas SM, Estep PW, Gore A, Hulick P, Labarga A, Lee JH, Lunshof JE, Kim BC, Kim JI, Li Z, Murray MF, Nilsen GB, Peters BA, Raman AM, Rienhoff HY, Robasky K, Wheeler MT. et al.A public resource facilitating clinical use of genomes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11920–11927. doi: 10.1073/pnas.1201904109. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
