

Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN
- PMID: 23539269
- PMCID: PMC3763966
- DOI: 10.1158/2159-8290.CD-12-0426
Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN
Abstract
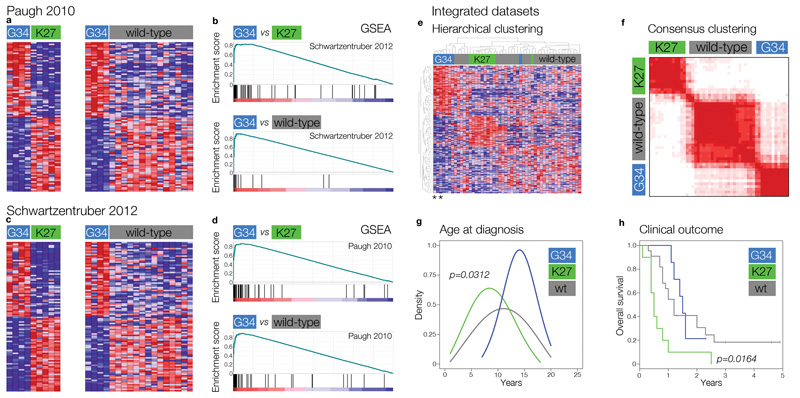
Children and young adults with glioblastoma (GBM) have a median survival rate of only 12 to 15 months, and these GBMs are clinically and biologically distinct from histologically similar cancers in older adults. They are defined by highly specific mutations in the gene encoding the histone H3.3 variant H3F3A , occurring either at or close to key residues marked by methylation for regulation of transcription—K27 and G34. Here, we show that the cerebral hemisphere-specific G34 mutation drives a distinct expression signature through differential genomic binding of the K36 trimethylation mark (H3K36me3). The transcriptional program induced recapitulates that of the developing forebrain, and involves numerous markers of stem-cell maintenance, cell-fate decisions, and self-renewal.Critically, H3F3A G34 mutations cause profound upregulation of MYCN , a potent oncogene that is causative of GBMs when expressed in the correct developmental context. This driving aberration is selectively targetable in this patient population through inhibiting kinases responsible for stabilization of the protein.
Significance: We provide the mechanistic explanation for how the fi rst histone gene mutation inhuman disease biology acts to deliver MYCN, a potent tumorigenic initiator, into a stem-cell compartment of the developing forebrain, selectively giving rise to incurable cerebral hemispheric GBM. Using synthetic lethal approaches to these mutant tumor cells provides a rational way to develop novel and highly selective treatment strategies
Figures



References
-
- Jones C, Perryman L, Hargrave D. Paediatric and adult malignant glioma: close relatives or distant cousins? Nature reviews. Clinical oncology. 2012;9:400–413. - PubMed
-
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. - PubMed
-
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3061–3068. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
