

The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin
- PMID: 23540698
- PMCID: PMC4035305
- DOI: 10.1016/j.cell.2013.02.033
The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin
Abstract
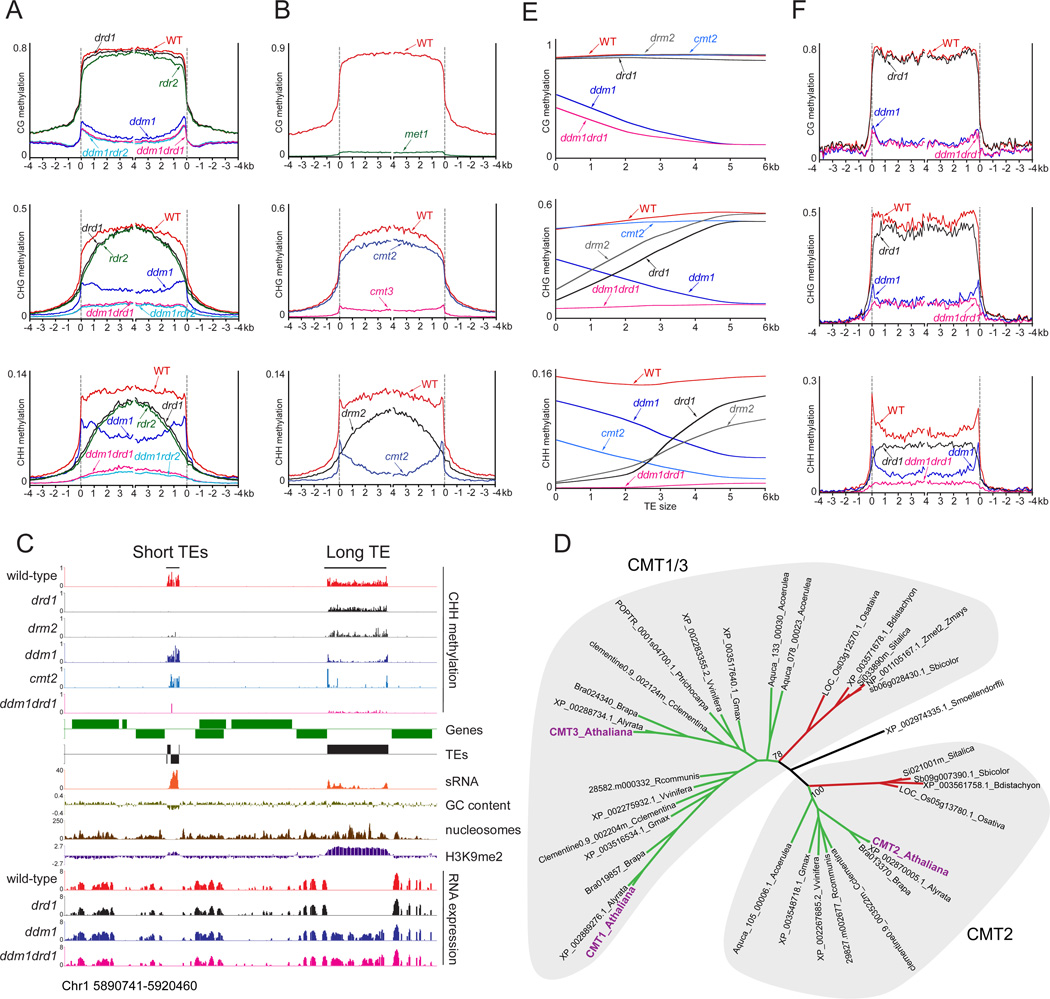
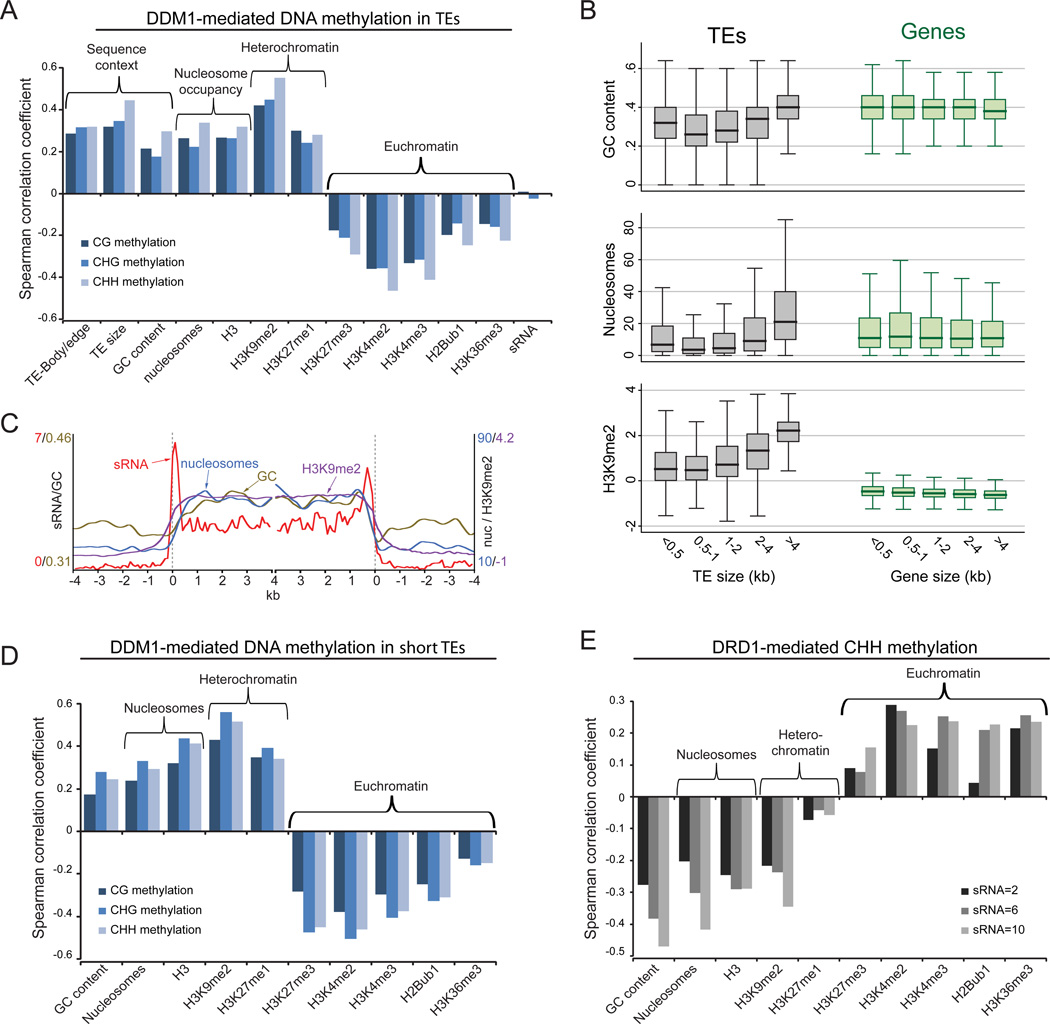
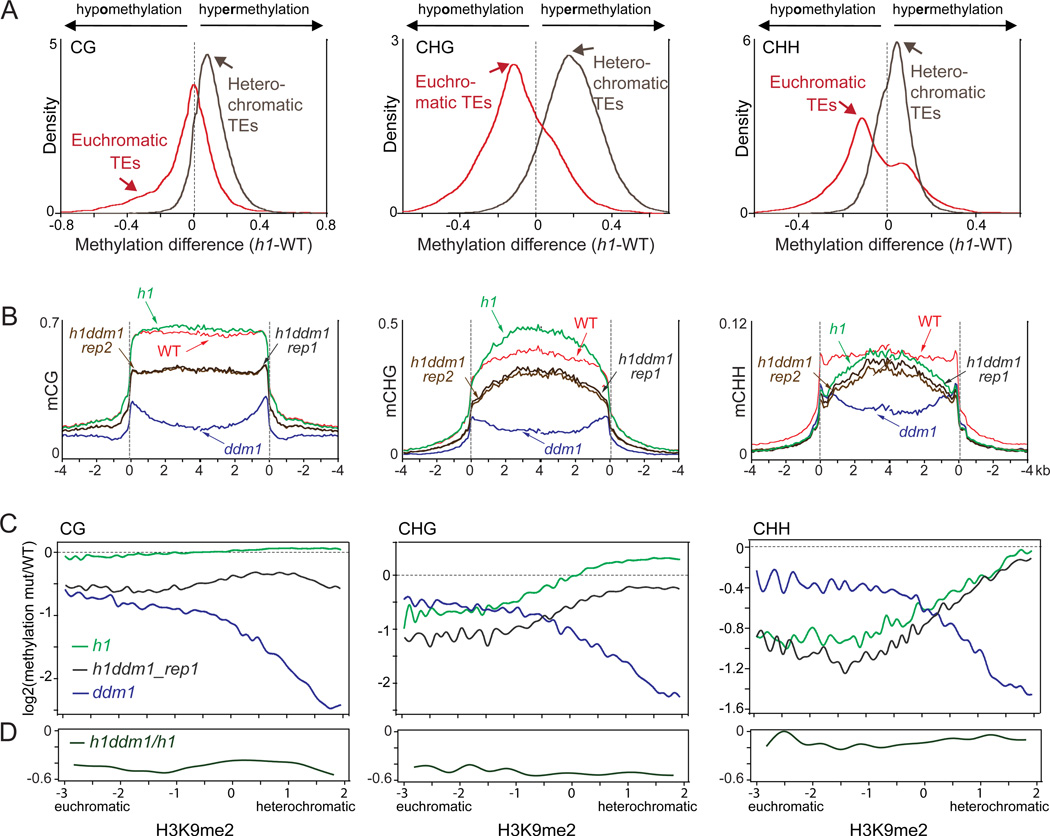
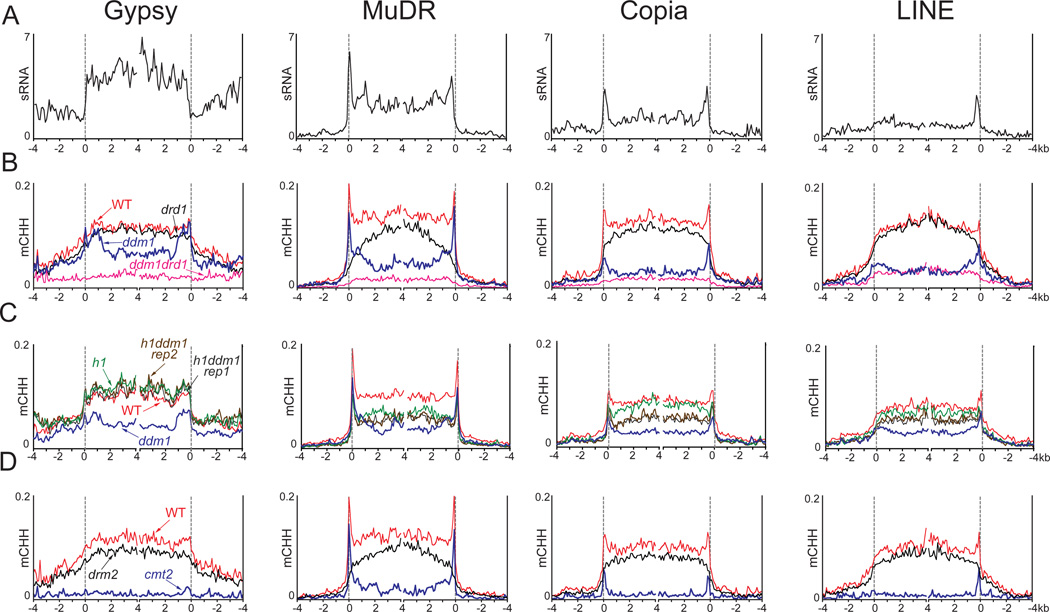
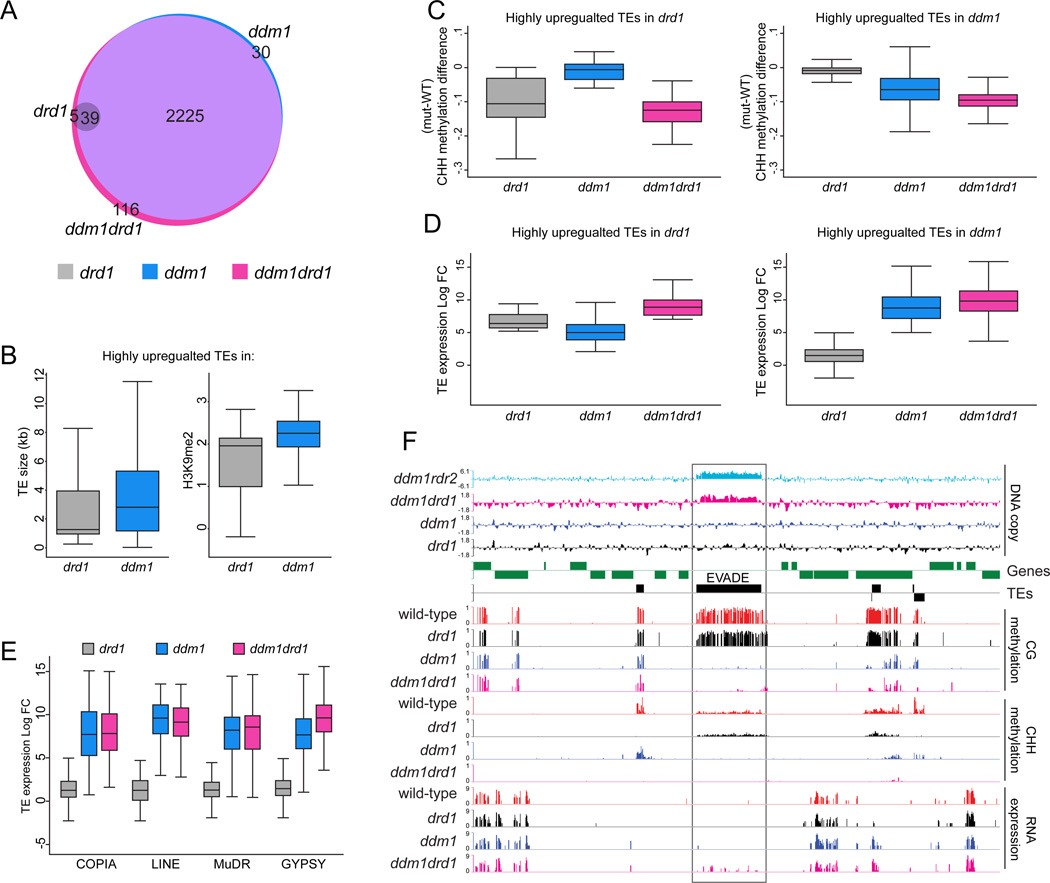
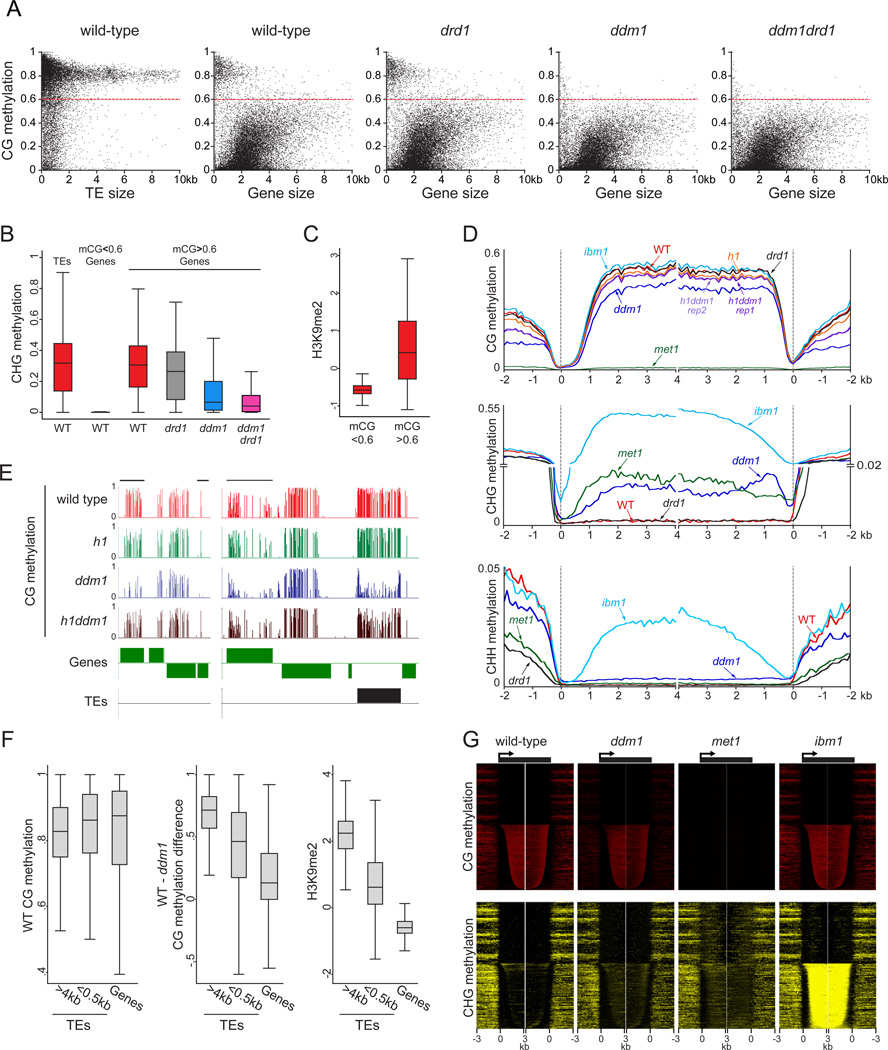
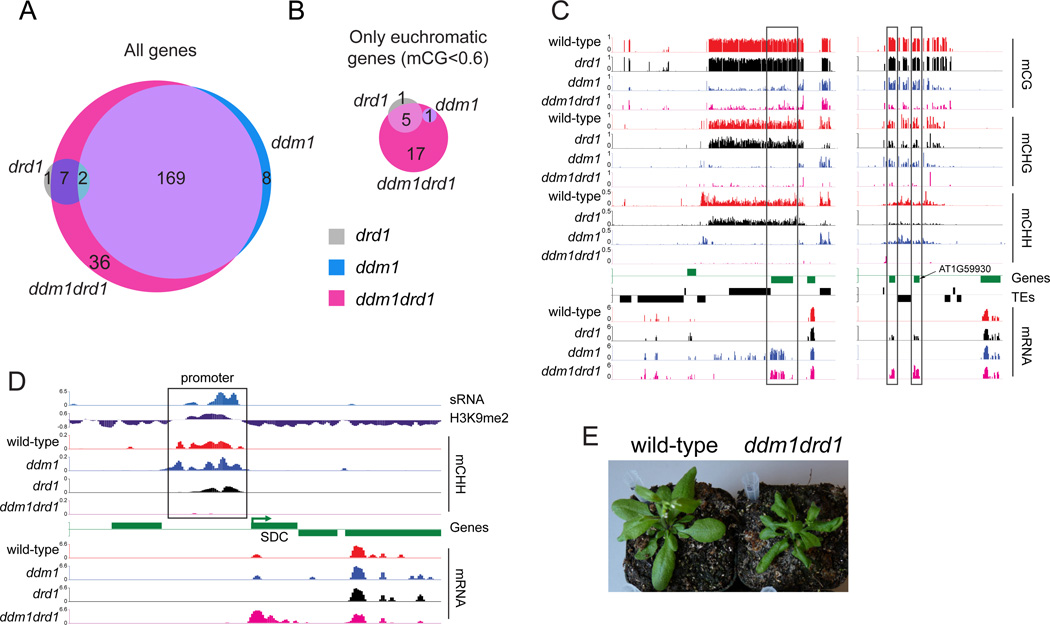
Nucleosome remodelers of the DDM1/Lsh family are required for DNA methylation of transposable elements, but the reason for this is unknown. How DDM1 interacts with other methylation pathways, such as small-RNA-directed DNA methylation (RdDM), which is thought to mediate plant asymmetric methylation through DRM enzymes, is also unclear. Here, we show that most asymmetric methylation is facilitated by DDM1 and mediated by the methyltransferase CMT2 separately from RdDM. We find that heterochromatic sequences preferentially require DDM1 for DNA methylation and that this preference depends on linker histone H1. RdDM is instead inhibited by heterochromatin and absolutely requires the nucleosome remodeler DRD1. Together, DDM1 and RdDM mediate nearly all transposon methylation and collaborate to repress transposition and regulate the methylation and expression of genes. Our results indicate that DDM1 provides DNA methyltransferases access to H1-containing heterochromatin to allow stable silencing of transposable elements in cooperation with the RdDM pathway.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Methylating the DNA of the most repressed: special access required.Mol Cell. 2013 Mar 28;49(6):1021-2. doi: 10.1016/j.molcel.2013.03.013. Mol Cell. 2013. PMID: 23541038 Free PMC article.
-
Reaching the inaccessible DNA.Nat Rev Mol Cell Biol. 2022 Jun;23(6):388. doi: 10.1038/s41580-022-00484-9. Nat Rev Mol Cell Biol. 2022. PMID: 35388188 No abstract available.
References
-
- Ascenzi R, Gantt JS. Subnuclear distribution of the entire complement of linker histone variants in Arabidopsis thaliana. Chromosoma. 1999;108:345–355. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
