

Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease
- PMID: 23541340
- PMCID: PMC3617381
- DOI: 10.1016/j.ajhg.2013.02.013
Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease
Abstract
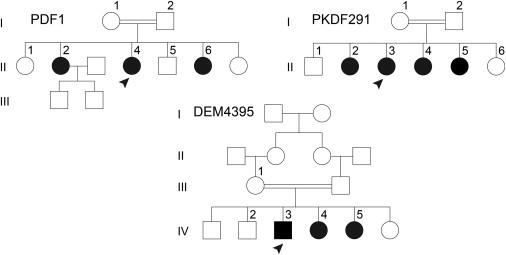
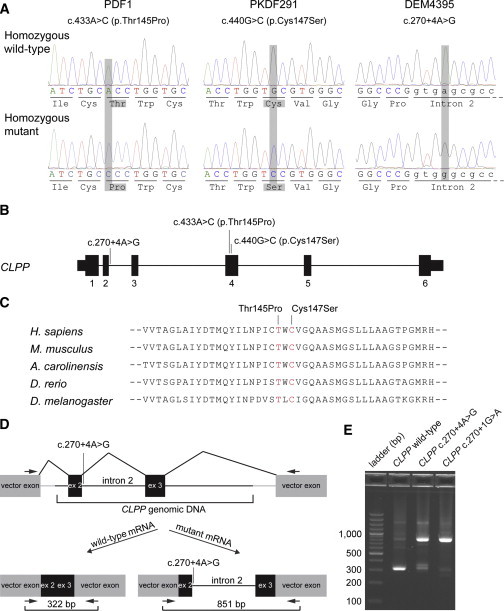
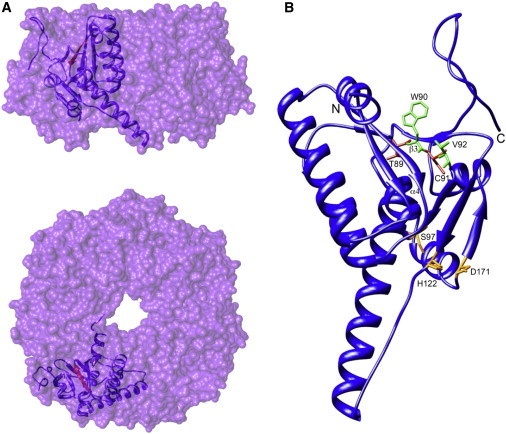
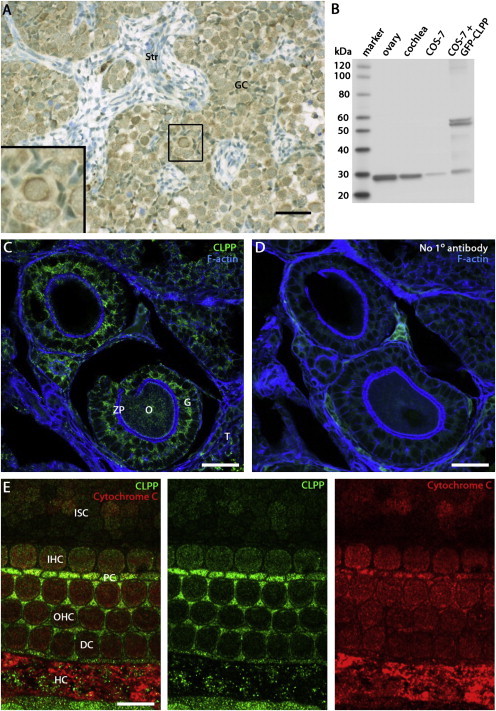
Perrault syndrome is a genetically and clinically heterogeneous autosomal-recessive condition characterized by sensorineural hearing loss and ovarian failure. By a combination of linkage analysis, homozygosity mapping, and exome sequencing in three families, we identified mutations in CLPP as the likely cause of this phenotype. In each family, affected individuals were homozygous for a different pathogenic CLPP allele: c.433A>C (p.Thr145Pro), c.440G>C (p.Cys147Ser), or an experimentally demonstrated splice-donor-site mutation, c.270+4A>G. CLPP, a component of a mitochondrial ATP-dependent proteolytic complex, is a highly conserved endopeptidase encoded by CLPP and forms an element of the evolutionarily ancient mitochondrial unfolded-protein response (UPR(mt)) stress signaling pathway. Crystal-structure modeling suggests that both substitutions would alter the structure of the CLPP barrel chamber that captures unfolded proteins and exposes them to proteolysis. Together with the previous identification of mutations in HARS2, encoding mitochondrial histidyl-tRNA synthetase, mutations in CLPP expose dysfunction of mitochondrial protein homeostasis as a cause of Perrault syndrome.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Guest S.S., Evans C.D., Winter R.M. The Online London Dysmorphology Database. Genet. Med. 1999;1:207–212. - PubMed
-
- Lenz D.R., Avraham K.B. Hereditary hearing loss: from human mutation to mechanism. Hear. Res. 2011;281:3–10. - PubMed
-
- Perrault M., Klotz B., Housset E. Deux cas de syndrome de Turner avec surdi-mutite dans une meme fratrie. Bull. Mem. Soc. Med. Hop. Paris. 1951;16:79–84. - PubMed
-
- Fiumara A., Sorge G., Toscano A., Parano E., Pavone L., Opitz J.M. Perrault syndrome: evidence for progressive nervous system involvement. Am. J. Med. Genet. A. 2004;128A:246–249. - PubMed
-
- Jenkinson E.M., Clayton-Smith J., Mehta S., Bennett C., Reardon W., Green A., Pearce S.H., De Michele G., Conway G.S., Cilliers D. Perrault syndrome: further evidence for genetic heterogeneity. J. Neurol. 2012;259:974–976. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- U54 HG006493/HG/NHGRI NIH HHS/United States
- R01 DC011651/DC/NIDCD NIH HHS/United States
- N01 HG065403/HG/NHGRI NIH HHS/United States
- R01 DC005641/DC/NIDCD NIH HHS/United States
- Z01 HL004232/ImNIH/Intramural NIH HHS/United States
- R01 DC003594/DC/NIDCD NIH HHS/United States
- T32 DC000039/DC/NIDCD NIH HHS/United States
- Z01 DC000039/ImNIH/Intramural NIH HHS/United States
- R01 CA157744/CA/NCI NIH HHS/United States
- K08 HL004232/HL/NHLBI NIH HHS/United States
- 088566/WT_/Wellcome Trust/United Kingdom
- DC000039-15/DC/NIDCD NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
