

Mutations in KCTD1 cause scalp-ear-nipple syndrome
- PMID: 23541344
- PMCID: PMC3617379
- DOI: 10.1016/j.ajhg.2013.03.002
Mutations in KCTD1 cause scalp-ear-nipple syndrome
Abstract
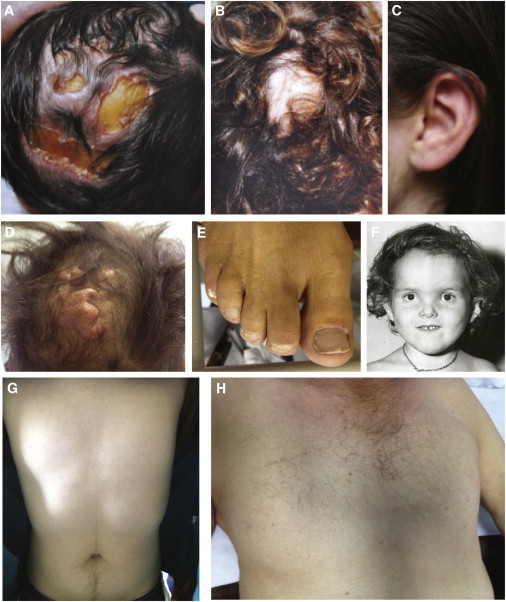
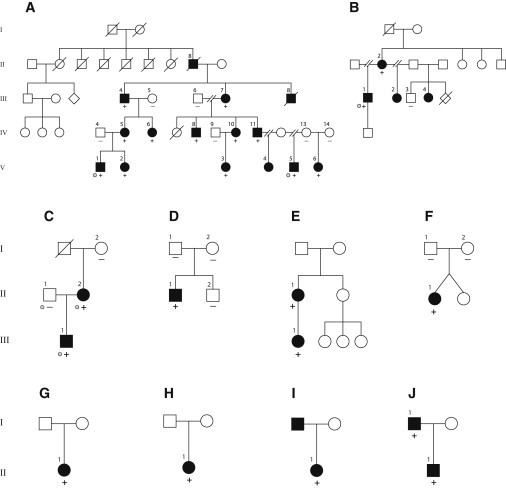
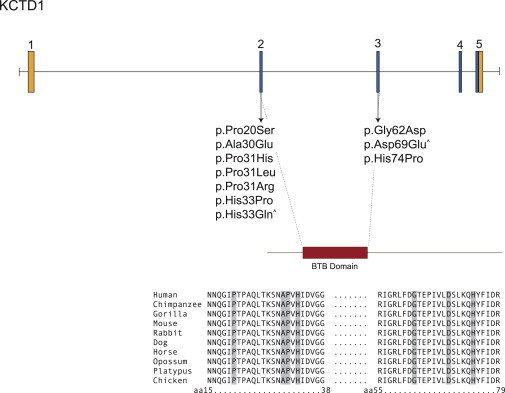
Scalp-ear-nipple (SEN) syndrome is a rare, autosomal-dominant disorder characterized by cutis aplasia of the scalp; minor anomalies of the external ears, digits, and nails; and malformations of the breast. We used linkage analysis and exome sequencing of a multiplex family affected by SEN syndrome to identify potassium-channel tetramerization-domain-containing 1 (KCTD1) mutations that cause SEN syndrome. Evaluation of a total of ten families affected by SEN syndrome revealed KCTD1 missense mutations in each family tested. All of the mutations occurred in a KCTD1 region encoding a highly conserved bric-a-brac, tram track, and broad complex (BTB) domain that is required for transcriptional repressor activity. KCTD1 inhibits the transactivation of the transcription factor AP-2α (TFAP2A) via its BTB domain, and mutations in TFAP2A cause cutis aplasia in individuals with branchiooculofacial syndrome (BOFS), suggesting a potential overlap in the pathogenesis of SEN syndrome and BOFS. The identification of KCTD1 mutations in SEN syndrome reveals a role for this BTB-domain-containing transcriptional repressor during ectodermal development.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Finlay A.Y., Marks R. An hereditary syndrome of lumpy scalp, odd ears and rudimentary nipples. Br. J. Dermatol. 1978;99:423–430. - PubMed
-
- Al-Gazali L., Nath R., Iram D., Al Malik H. Hypotonia, developmental delay and features of scalp-ear-nipple syndrome in an inbred Arab family. Clin. Dysmorphol. 2007;16:105–107. - PubMed
-
- Baris H., Tan W.H., Kimonis V.E. Hypothelia, syndactyly, and ear malformation—a variant of the scalp-ear-nipple syndrome?: Case report and review of the literature. Am. J. Med. Genet. A. 2005;134A:220–222. - PubMed
-
- Edwards M.J., McDonald D., Moore P., Rae J. Scalp-ear-nipple syndrome: additional manifestations. Am. J. Med. Genet. 1994;50:247–250. - PubMed
-
- Naik P., Kini P., Chopra D., Gupta Y. Finlay-Marks syndrome: report of two siblings and review of literature. Am. J. Med. Genet. A. 2012;158A:1696–1701. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- K99 HG004316/HG/NHGRI NIH HHS/United States
- U54 HG006493/HG/NHGRI NIH HHS/United States
- RC2 HG005608/HG/NHGRI NIH HHS/United States
- U54 HG006542/HG/NHGRI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- 1U54HG006493/HG/NHGRI NIH HHS/United States
- 5R01HG004316/HG/NHGRI NIH HHS/United States
- 1RC2HG005608/HG/NHGRI NIH HHS/United States
- R00 HG004316/HG/NHGRI NIH HHS/United States
- K23 HD057331/HD/NICHD NIH HHS/United States
- R01 HD048895/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
