

Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors
- PMID: 23542178
- PMCID: PMC3982880
- DOI: 10.1038/onc.2013.92
Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors
Abstract
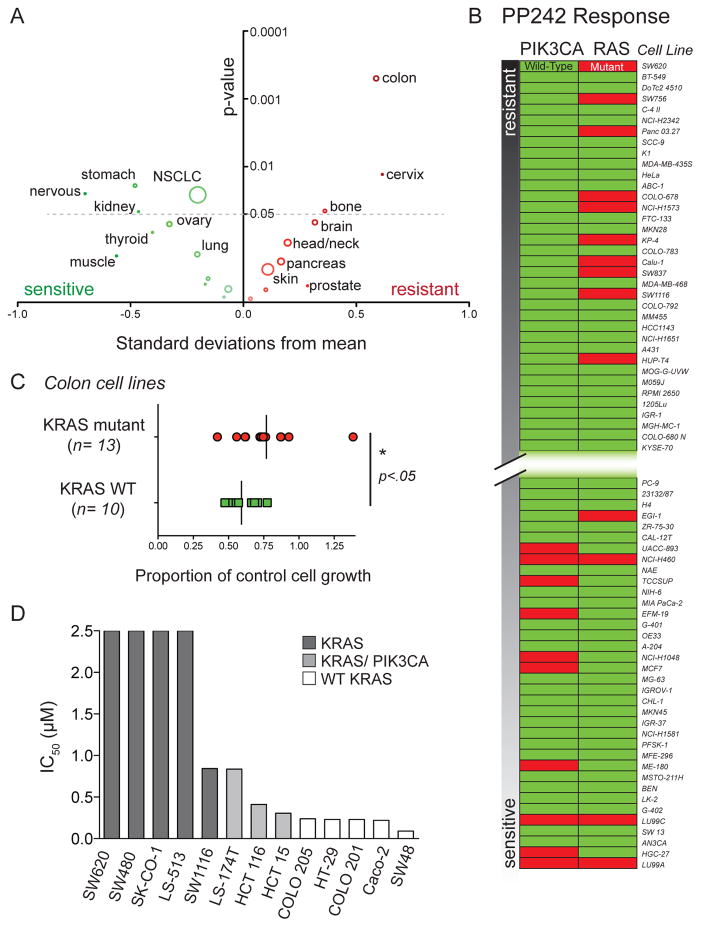
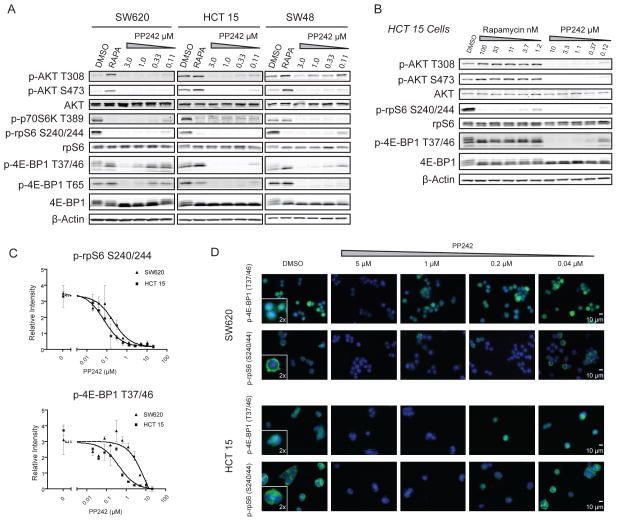
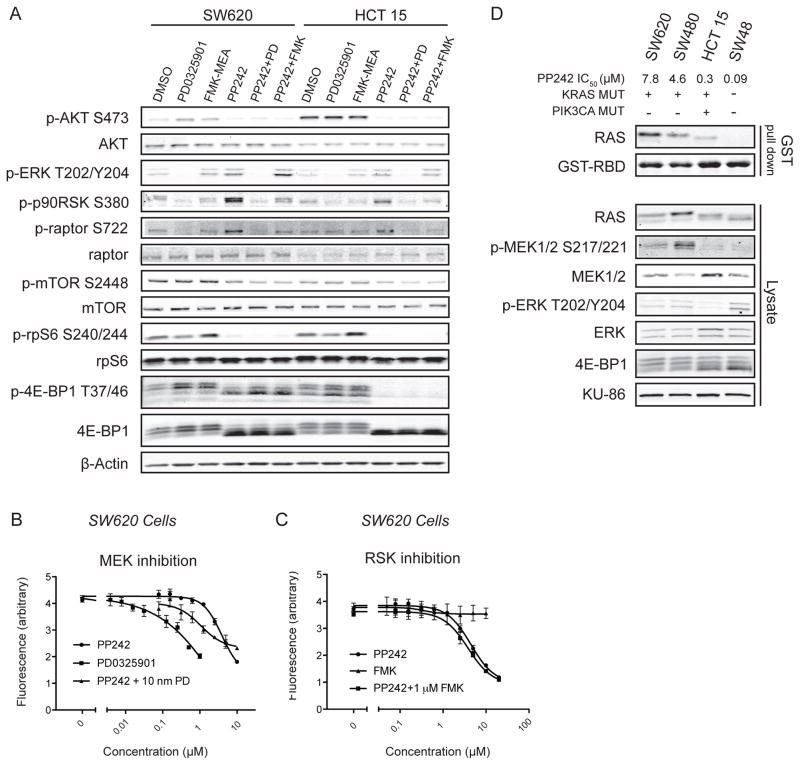
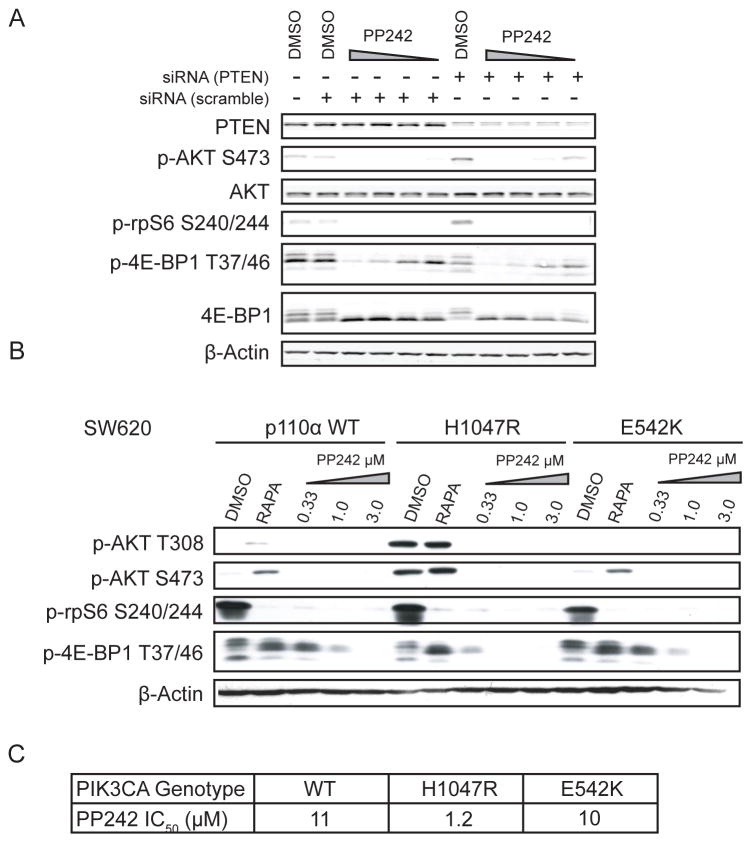
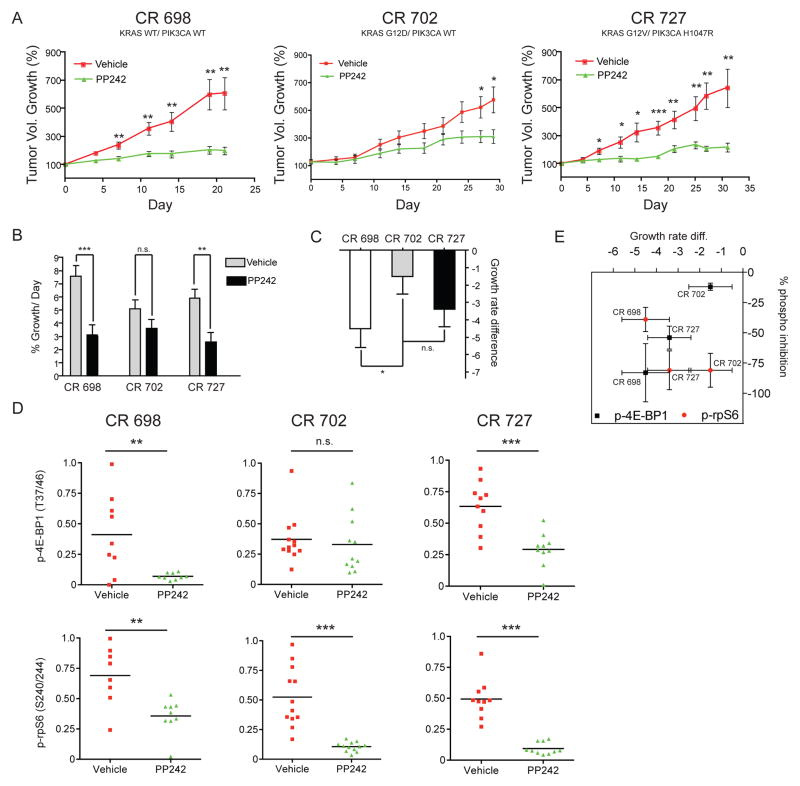
The mammalian target of rapamycin (mTOR) regulates cell growth by integrating nutrient and growth factor signaling and is strongly implicated in cancer. But mTOR is not an oncogene, and which tumors will be resistant or sensitive to new adenosine triphosphate (ATP) competitive mTOR inhibitors now in clinical trials remains unknown. We screened a panel of over 600 human cancer cell lines to identify markers of resistance and sensitivity to the mTOR inhibitor PP242. RAS and phosphatidylinositol 3-kinase catalytic subunit alpha (PIK3CA) mutations were the most significant genetic markers for resistance and sensitivity to PP242, respectively; colon origin was the most significant marker for resistance based on tissue type. Among colon cancer cell lines, those with KRAS mutations were most resistant to PP242, whereas those without KRAS mutations most sensitive. Surprisingly, cell lines with co-mutation of PIK3CA and KRAS had intermediate sensitivity. Immunoblot analysis of the signaling targets downstream of mTOR revealed that the degree of cellular growth inhibition induced by PP242 was correlated with inhibition of phosphorylation of the translational repressor eIF4E-binding protein 1 (4E-BP1), but not ribosomal protein S6 (rpS6). In a tumor growth inhibition trial of PP242 in patient-derived colon cancer xenografts, resistance to PP242-induced inhibition of 4E-BP1 phosphorylation and xenograft growth was again observed in KRAS mutant tumors without PIK3CA co-mutation, compared with KRAS wild-type controls. We show that, in the absence of PIK3CA co-mutation, KRAS mutations are associated with resistance to PP242 and that this is specifically linked to changes in the level of phosphorylation of 4E-BP1.
Conflict of interest statement
Figures
References
-
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002 Feb 28;346(9):645–52. - PubMed
-
- Jänne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009 Mar 8;8(9):709–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
