

doi: 10.1172/JCI61398.
Epub 2013 Apr 1.
A mitochondrial bioenergetic etiology of disease
Affiliations
- PMID: 23543062
- PMCID: PMC3614529
- DOI: 10.1172/JCI61398
Item in Clipboard
A mitochondrial bioenergetic etiology of disease
J Clin Invest.
2013 Apr.
Abstract
The classical Mendelian genetic perspective has failed to adequately explain the biology and genetics of common metabolic and degenerative diseases. This is because these diseases are primarily systemic bioenergetic diseases, and the most important energy genes are located in the cytoplasmic mitochondrial DNA (mtDNA). Therefore, to understand these "complex" diseases, we must investigate their bioenergetic pathophysiology and consider the genetics of the thousands of copies of maternally inherited mtDNA, the more than 1,000 nuclear DNA (nDNA) bioenergetic genes, and the epigenomic and signal transduction systems that coordinate these dispersed elements of the mitochondrial genome.
Figures
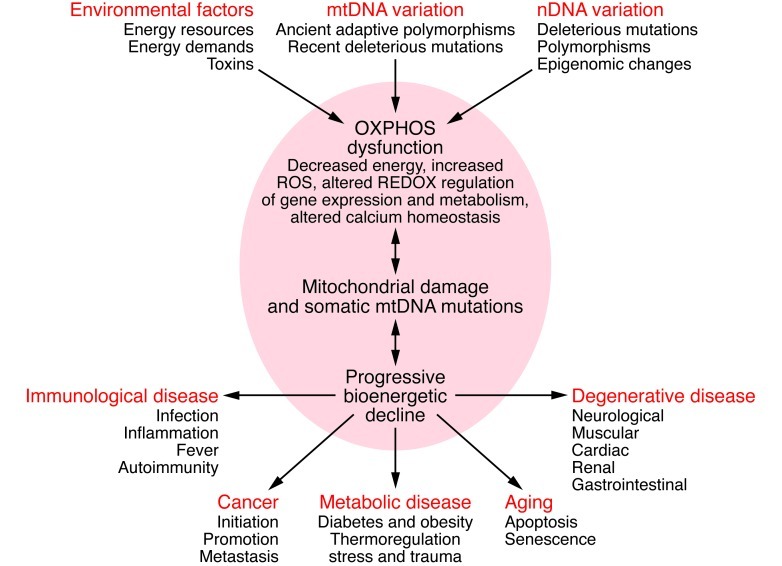
Mitochondrial OXPHOS can be perturbed by nDNA genetic alterations and/or epigenomic regulation, by mtDNA ancient adaptive of recent deleterious mutations, or by variation in the availability of calories and in caloric demands. Alterations in mitochondrial structure and function can impair OXPHOS, which in turn can reduce energy production, alter cellular redox state, increase ROS production, deregulate Ca2+ homeostasis, and ultimately activate the mtPTP, leading to apoptosis. These and other consequences of OXPHOS perturbation can destabilize mtDNA. This results in progressive accumulation of somatic mtDNA mutations and decline of mitochondrial function, which accounts for aging and the delayed-onset and progressive course of degenerative diseases. As energy output declines, the most energetic tissues are preferentially affected, resulting in degenerative diseases of the central nervous system, heart, muscle, and kidney. Aberrant mitochondrial caloric metabolism also leads to metabolic deregulation, endocrine dysfunction, and symptoms such as diabetes, obesity, and cardiovascular disease. The release into the blood stream of mtDNA mutant N-formylmethionine polypeptides plus the mtDNA can initiate the inflammatory response, contributing to autoimmune diseases (e.g., multiple sclerosis and type I diabetes) and possibly also to the inflammatory component of late-onset degenerative diseases. Finally, cancer cells must manage energy resources to permit rapid replication (95). Figure adapted with permission from Cold Spring Harbor Press (55).
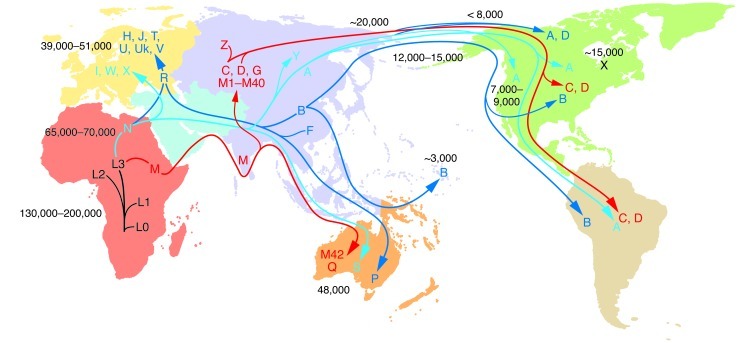
The uniparentally inherited mtDNA can only change by sequential accumulation of mutations along radiating female lineages. Therefore, the mtDNA mutational tree and ancient migrations of women were reconstructed by sequencing mtDNAs from indigenous populations and correlating their regional clusters of related haplotypes (haplogroups) with the population’s geographic location. The haplogroups are regional because they were founded by regionally adaptive functional variants. The mtDNA tree originates in Africa, and all African mtDNAs are classified together as macrohaplogroup L. From haplogroup L3, two mtDNA lineages, M and N, arose in Ethiopia and successfully left Africa to colonize the rest of the world about 65,000–70,000 YBP. The founder mtDNA of macrohaplogroup N harbored two mtDNA missense mutations, ND3 G10398A (A114T) and ATP6 G8701A (A59T), whereas the founder of macrohaplogroup M did not harbor major functional mutations (3, 15). Early M and N emigrants from Africa moved through Southeast Asia, ending in Australia (96, 97). N mtDNAs also moved north from Africa into the Middle East to generate submacrohaplogroup R and European-specific haplogroups H, J, T, U, Uk, and V (from R) and I, W, and X (from N). N and R gave rise to Asian haplogroups A+Y and B+F, respectively. M moved north out of Southeast Asia to colonize Asia, generating haplogroups C and D and multiple M haplogroups. Haplogroups A, B, C, D, and X subsequently migrated to the Americas. The mtDNA mutation rate is 2.2%–2.9% per million years (numbers within the figure denote YBP). Figure adapted with permission from MITOMAP (5).
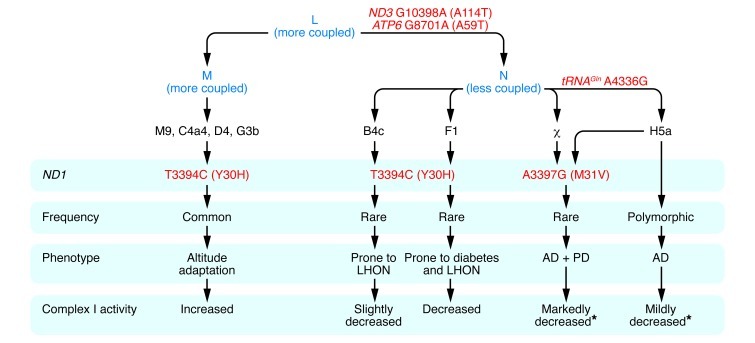
African haplogroup L3 gave rise to macrohaplogroups M and N, which colonized Europe and Asia. N differed from M in harboring the ND3 G10398A (A114T) and ATP6 G8701A (A59T) variants. In Europe, N gave rise to haplogroup H, and H acquired the tRNAGln A4336G variant to generate H5a, which predisposes to AD, PD, and both AD and PD (17). The ND1 A3397G (M31V) missense mutation arose twice in Europeans, once on H5a and once independently, and in both cases was associated with predisposition to both AD and PD (17). Both tRNAGln A4336G and ND1 A3397G (M31V) mutations are likely to reduce mitochondrial complex I activity, augmenting the founding N variants. The ND1 T3394C (Y30H) mutation, which is adjacent to the ND1 M31 codon, arose on N and M mtDNAs. When arising on N haplogroups B and F, the ND1 T3394C (Y30H) variant is associated with complex I deficiency and increased penetrance of the primary LHON mutations. However, complex I activity is also modulated by N haplogroup background, with haplogroup F mtDNAs having lower complex I activity than haplogroup B mtDNAs, consistent with haplogroup F predisposition to diabetes (23). The ND1 T3394C (Y30H) mutation has arisen on several M mtDNAs, with all haplogroup M9 mtDNAs having the 3394C allele. Both M9 and 3394C mtDNAs increase in frequency with altitude in Tibet. Finally, M9 complex I activity is equal to or greater than that of any of the N haplogroups with the wild-type T3394 allele (20). Asterisks indicate that the stated complex I activity is predicted, based on the known genotype and complex I activities determined for cell lines harboring ND1 T3394C (Y30H)–containing mtDNAs.
References
-
- Wallace DC, Lott MT, Procaccio V. Mitochondrial medicine: the mitochondrial biology and genetics of metabolic and degenerative diseases, cancer, and aging. In: Rimoin DL, Pyeritz RE, Korf BR, eds.Emery and Rimoin’s Principles and Practice of Medical Genetics. Philadelphia, Pennsylvania, USA: Churchill Livingstone Elsevier; 2013.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
