

SeqEntropy: genome-wide assessment of repeats for short read sequencing
- PMID: 23544073
- PMCID: PMC3609794
- DOI: 10.1371/journal.pone.0059484
SeqEntropy: genome-wide assessment of repeats for short read sequencing
Abstract
Background: Recent studies on genome assembly from short-read sequencing data reported the limitation of this technology to reconstruct the entire genome even at very high depth coverage. We investigated the limitation from the perspective of information theory to evaluate the effect of repeats on short-read genome assembly using idealized (error-free) reads at different lengths.
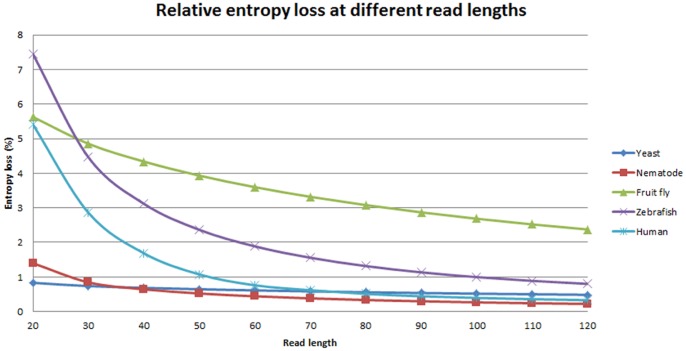
Methodology/principal findings: We define a metric H(k) to be the entropy of sequencing reads at a read length k and use the relative loss of entropy ΔH(k) to measure the impact of repeats for the reconstruction of whole-genome from sequences of length k. In our experiments, we found that entropy loss correlates well with de-novo assembly coverage of a genome, and a score of ΔH(k)>1% indicates a severe loss in genome reconstruction fidelity. The minimal read lengths to achieve ΔH(k)<1% are different for various organisms and are independent of the genome size. For example, in order to meet the threshold of ΔH(k)<1%, a read length of 60 bp is needed for the sequencing of human genome (3.2 10(9) bp) and 320 bp for the sequencing of fruit fly (1.8×10(8) bp). We also calculated the ΔH(k) scores for 2725 prokaryotic chromosomes and plasmids at several read lengths. Our results indicate that the levels of repeats in different genomes are diverse and the entropy of sequencing reads provides a measurement for the repeat structures.
Conclusions/significance: The proposed entropy-based measurement, which can be calculated in seconds to minutes in most cases, provides a rapid quantitative evaluation on the limitation of idealized short-read genome sequencing. Moreover, the calculation can be parallelized to scale up to large euakryotic genomes. This approach may be useful to tune the sequencing parameters to achieve better genome assemblies when a closely related genome is already available.
Conflict of interest statement
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
